蛋白质结构预测的突破
导语
了解一个生物过程所需的关键信息之一是其组成蛋白的结构,但结构测定的实验方法往往耗时费力,而且结果不确定,需要投入大量的时间和资源。相比之下,蛋白质序列很容易通过翻译基因组序列获得,并且可以获得大量的蛋白质。由于蛋白质的结构是由其序列决定,因此试图从另一个序列中推导出蛋白质的折叠问题–已经持续了半个世纪,其重要性随着序列数据库的指数增长而上升,并对连续的方法未能带来决定性的进展感到沮丧。事实上,从本世纪的第一个十年开始,蛋白质科学界越来越意识到这个问题是计算生物学的巨大挑战之一。

DrugAI | 来源

事情并不是这样开始的。莱纳斯-鲍林从多肽链的立体化学考虑建立的二级结构模型,以及不久之后证明这种二级结构可以被组装成α-角蛋白和胶原蛋白的三维模型,导致人们期待几何考虑、模型建立和参数方程的结合可以解决蛋白质结构的原理,正如他们已经为核酸做的那样。然而,第一个蛋白质晶体结构及其惊人的不规则性使人们认识到,这些原理可能比预期的要复杂得多。
尽管如此,在20世纪90年代初,人们对通过简化多肽链的生物物理表征和线程所取得的进展感到兴奋,认为在从氨基酸序列推断结构方面取得了快速、决定性的进展。然而,这些方法在现实生活中的应用与此并不匹配,而且很明显,一些报告的成功可能是由于 “后预测”,即预测者已经知道其结构的目标。为了获得对蛋白质结构预测技术水平的客观评估,由马里兰大学的John Moult领导的一组科学家在1994年组织了一次实验,即CASP(结构预测的关键评估),预测者可以在一个双盲框架内评估他们的方法。组织者将收集结构尚未公布(在某些情况下甚至尚未完全确定)的蛋白质序列,并将其作为预测目标提供给计算科学家。然后,组织者将把提交的预测和已解决的结构交给评估者,评估者对参与预测的团队不了解,只知道组号。在实验结束时(每两年重复一次),将举行一次会议,讨论结果。
CASP1是一个令人清醒的经验,因为结构预测的工具被证明是非常钝的。用组织者的话说:”这些预测出了很多问题,这也是实验的主要价值所在。一个可靠的信息来源是与目标蛋白相关的结构,具有已知结构的亲属的目标被归类为最简单的,可以通过同源模板上的建模获得。然而,由于检测和目标与模板比对的错误,要建立一个比最近的可用模板更接近目标的模型是相当困难的。应用生物物理方法,如能量最小化,似乎只会使错误更严重。相应地,CASP2增加了对更远的同源物的检测、建模和细化的投入。CASP2比CASP1更成功,特别是在CASP1预测基本上是随机的、结构数据库中缺乏可检测模板的较难目标,但进展仍然有限。《纽约时报》以 “蛋白质1,计算机0 “作为著名的报道标题,并引用了一位组织者的话,他看到了一些小的进展迹象,”这是令人鼓舞的,但离有用的东西还有很长的路要走”,而一位评估员则夸奖说 “不能再保证失败了”。
在此基础上,CASP3-5实验提供了进一步的改进,然而,主要是在中等难度的靶标领域,通过一系列日益强大的序列搜索工具,可以检测到更加遥远的同源物。事实证明,同源蛋白基本上保持着相同的折叠,即使它们的序列似乎已经分化到了不相似的 “午夜区”。相比之下,将生物物理参数纳入预测方法的努力,虽然为较小的靶标提供了一些令人印象深刻的成功,但并没有扩展到较大的靶标,使进化相关度的统计检测成为结构预测的主要工具。
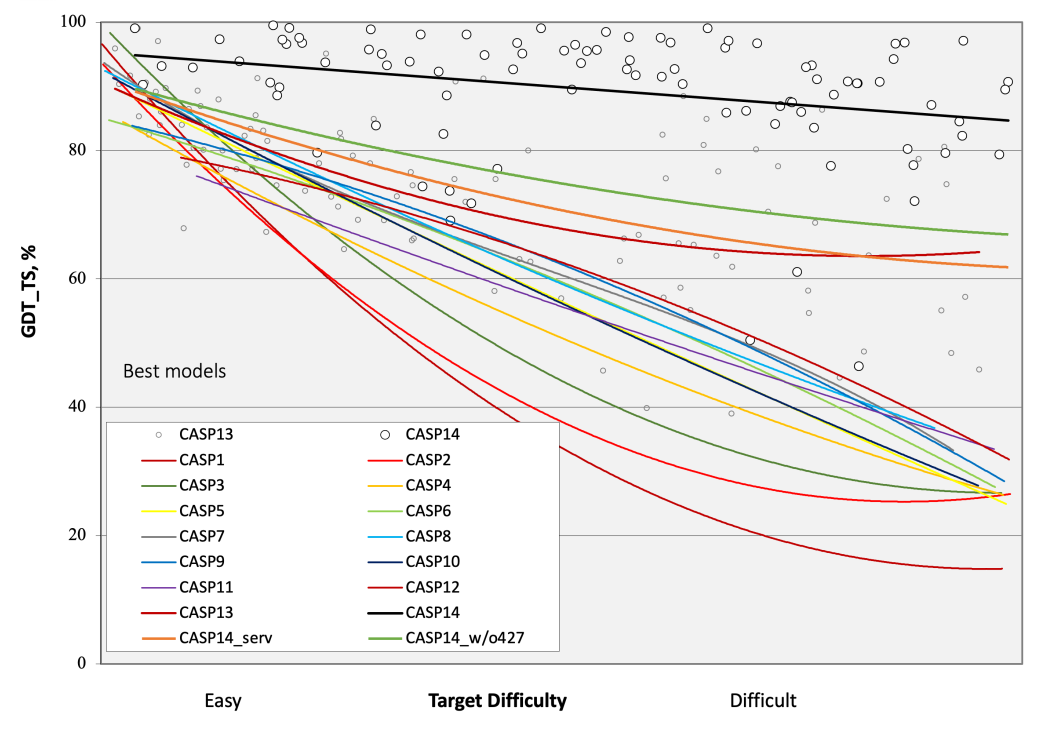
CASP3中引入的模型准确性测量方法GDT-TS(全局距离测试-总分),可以比较实验内部和实验之间的结果。在对两个具有相同序列的结构进行迭代叠加后,该方法比较了同源的Cα碳的位置,统计了在1、2、4和8Å的距离分界线内的配对百分比,将百分比相加并除以4。这种对相似性的关注使该测量方法能够将那些差的、但包含局部正确片段的模型与全局错误的模型区分开来,这是其他相关测量方法(如均方根偏差)所不能实现的。非常粗略的说,GDT-TS的得分在20分左右,表示大体上是随机的模型,得分在50分左右,具有整体正确拓扑结构的模型,得分在70分左右,具有准确的全局和局部拓扑结构的模型。超过80分,结构细节的建模越来越正确,超过95分,模型就像根据实验数据建立的模型一样准确。
John Moult在CASP14会议的介绍中提出了CASP单个实验中GDT-TS得分的概述,显示为通过该实验中每个目标取得的最佳得分的多项式拟合(https://en.wikipedia.org/wiki/AlphaFold#/media/File:CASP_results_2020.png)。这一概述表明,在CASP5之后,整体进展基本上停滞不前,直到CASP12,导致该领域的一些人怀疑我们是否会得到问题的解决。然而,下一步进展的种子已经播下。
从CASP实验开始,科学家们就想知道是否有可能从同源蛋白质的多序列比对中的相关突变计算出残基间的接触图,以获得折叠的指纹并指导结构预测。研究人员的想法是,如果突变是相关的,那么这些位置的残基很可能是物理接触的,提供的信息可以通过核磁共振确定结构。然而,多年来,这种接触图的准确性仍然很低,因为以成对的方式分析相关性,无法区分直接的、结构性的相关性和间接的、功能性的相关性。从2010年左右开始,通过使用直接耦合分析进行全局接触预测,可以更好地区分不同的共同演化的残基,该方法同时考虑了所有成对的相互作用,并根据观察到的成对的相关性对接触图进行全局优化。这种方法在2017年又向前迈进了一大步,证明了深度学习方法不仅可以通过这种方式从多个排列中提取高质量的接触图,甚至在同源物很少的情况下也可以将预测的接触解释为一组距离,从而为基础折叠提供更精细的几何指纹。卷积神经网络在距离图预测中的应用被领先的结构预测小组在CASP13(2018)中使用,并对硬产生了强大的影响,对于这些目标,最佳模型的GDT-TS从40左右上升到60以上。
在CASP13的高分小组中,有一个出乎意料的新成员AlphaFold,由Alphabet公司的领先人工智能实验室DeepMind派出。令所有人惊讶的是,这个小组以其关键的洞察力击败了所有参赛者,即距离图的概率分布可以转换为特定蛋白质的统计潜力,从而通过最小化生成蛋白质折叠。虽然AlphaFold在CASP13中的领先优势比以往CASP实验中排名第一和第二的小组之间的典型距离要大,但它的总体表现是递增性的,而不是变革性的,只在大约三分之一的情况下提供了最佳模型,尽管对较难的目标的领先优势大于对较易的目标的领先优势。
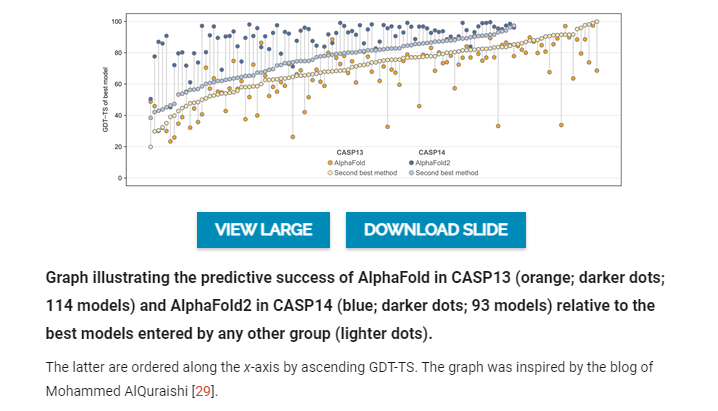
因此,没有人准备好迎接AlphaFold的第二个化身AlphaFold2在CASP14上的变革性表现,它远远领先于所有其他参与者,其预测的GDT-TS中值达到了92.4! 回顾一下,这是在实验结构的范围内,导致许多人得出结论,单条蛋白质链的结构预测问题现在已经解决了,正如John Moult在CASP14会议的总结发言中所说。将AlphaFold2的预测结果与其他研究小组提交的最佳模型进行比较,可以清楚地看到进步的程度,因为AlphaFold2的预测结果通常是GDT-TS得分>80,即使是最难的目标,而这些目标的第二好的模型都低于60。
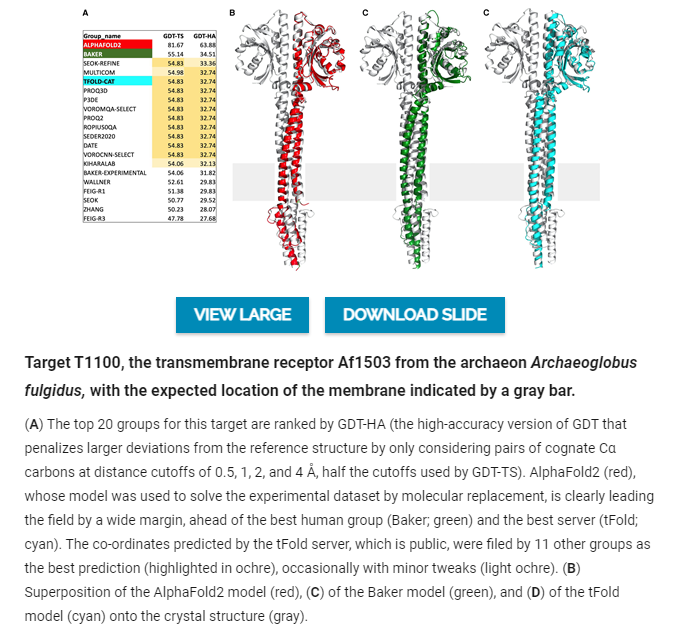
为了说明这一点,简单介绍一下目标T1100的情况,这是一个古生物跨膜受体,AlphaFold2为其提交了一个GDT-TS约为80的模型,而接下来最好的一组模型的GDT-TS约为55。研究人员的小组进入这个目标是由于2020年8月组织者和评审员的一次在线会议,在这次会议上,评审员之一Nick Grishin将427小组(后来发现是AlphaFold2)的惊人预测简洁地提到了一个点上。所以,要么这个小组接近解决折叠问题,要么他们以某种方式作弊”。作为回应,研究者提到有一个跨膜受体的衍射数据,由于相位问题,几乎十年都没有解决这个问题。427组文件模型是否足以通过分子置换解决该数据集?当然,这是不可能作弊的。简而言之,用AlphaFold2模型可以很容易地解决这个结构。其他提交的模型有很好的整体拓扑结构,但有很多地方偏离了结构,使它们成为分子置换的不良模板。作为一个有趣的侧面,在这个20个排名最高的小组中,有12个server提交了一个公共预测服务器的预测坐标作为他们的最佳答案,偶尔也有一些细化的尝试。其中一个服务器名为tFold,由中国科技公司腾讯的人工智能实验室运营,这表明DeepMind并不是唯一有兴趣加入这一战局的公司实验室。
是什么让AlphaFold2建立了这种领先优势?更详细的评估要等CASP14论文集中的方法发表后才能进行,但从John Jumper代表AlphaFold2团队在CASP14会议上的发言以及该领域专家的意见来看,预测网络的架构已经发生了根本性的变化。AlphaFold使用卷积神经网络进行距离图预测,并应用梯度下降优化法从这些约束条件中构建模型,而AlphaFold2构建了一个端到端的网络,从序列输入到结构输出,模型参数可以共同调整,以优化最终模型,而不是沿途的代理措施。这种用于网络优化的端到端训练是由Mohammed Al Quraishi在CASP13之后提出的,在这里被证明是预测成功的一个重要组成部分。此外,AlphaFold2使用注意力模块来推导距离约束,并通过三维等价变换器神经网络从中建立结构模型,该网络直接在三维空间的原子上操作。起源于自然语言处理的注意力模块并不从输入的多序列排列中得出汇总统计,而是选择一个序列子集来关注,并得出第一个距离图,在此基础上决定在下一次迭代中关注哪些序列。这样,通过迭代优化,该网络甚至可以从含有少量全长同源物的序列排列中提取更丰富的约束条件,这也是其相对于所有其他方法在硬目标上的表现尤为突出的原因。这个网络结构的总体战略似乎是以最佳的局部解决方案为目标,以便从这些解决方案中组合出全局模型,这显然是非常成功的。
那么,DeepMind解决了蛋白质折叠问题吗?就其基本形式而言–从一个蛋白质的氨基酸序列推导出它的原生结构–CASP14的答案对大多数蛋白质来说似乎是肯定的,只要程序能够访问蛋白质序列和结构数据库,并且目标蛋白质是折叠的。在研究人员看来,关于解决方案意味着理解或预测不是从单一的氨基酸序列进行的反对意见归结为语义学。然而,蛋白质折叠问题比仅仅从序列中推断出静态三维结构要复杂得多。一个蛋白质序列不仅包含了结构的信息,而且还包含了达到这个结构的路径,以及它在应对不断变化的条件和结合伙伴时所经历的动态调整,以及它需要参与的细胞机器的组件,以达到其原生位置。从其序列中的信息,一个蛋白质可以识别其结合伙伴,并知道它是否会通过催化或构象变化来改变这些伙伴,以及它是否会在遇到它们时有条件地折叠或展开。所有这些方面,目前都不在AlphaFold2的范围内,但对于蛋白质的生物功能来说是至关重要的,科学家们对这些方面最感兴趣是可以理解的。因此,研究人员会得出这样的结论:不,AlphaFold2不是解决蛋白质折叠问题的最后一步,而是在实现蛋白质结构预测目标的一条非常令人兴奋的新道路上迈出的第一步,现在可能已经触手可及了。
这是否意味着AlphaFold2获得的进步被夸大了,事实上并不尽如人意?对这一点也肯定是否定的。研究人员发现,这一进展绝对是令人震惊的,这一点我们在CASP14的媒体报道中反复强调。研究人员认为,实现这一突破的漫长而艰辛的历程,涉及到生物物理学和计算生物学中一些最聪明的头脑,充分证明了这一成就的巨大。事实上,这一进展需要引入深度学习方法,这促使我们问道,结构预测问题对于人类的大脑来说是否太难解决。套用J.B.S. Haldane的话,他怀疑宇宙不仅比我们想象的要奇怪,而且比我们能想象的要奇怪,这个问题可能比我们能解决的要难?
研究人员担心情况确实如此,端到端训练成功的原因之一是消除了人类的偏见。训练有素的科学家数十年的努力和许多数十亿美元的公共投资显然产生了突破问题所需的数据,但这一突破需要计算网络,而计算网络与人脑不同,是为分析非线性相关关系而优化。就像许多其他群体一样–运动员和国际象棋选手就是其中之一–我们将不得不习惯于机器拥有超出我们生物范围的能力这一事实。我们期待着我们认为将是一波先进的预测服务器,既来自领先的学术团体,也来自具有先进机器学习能力的公司,这将使蛋白质的结构空间像25年前BLAST对序列空间所做的那样广泛而迅速地被访问,标志着生命科学的类似革命。
参考资料:
Andrei N. Lupas, Joana Pereira, Vikram Alva, Felipe Merino, Murray Coles, Marcus D. Hartmann; The breakthrough in protein structure prediction. Biochem J 28 May 2021; 478 (10): 1885–1890.
doi: https://doi.org/10.1042/BCJ20200963
复杂科学最新论文
集智斑图顶刊论文速递栏目上线以来,持续收录来自Nature、Science等顶刊的最新论文,追踪复杂系统、网络科学、计算社会科学等领域的前沿进展。现在正式推出订阅功能,每周通过微信服务号「集智斑图」推送论文信息。扫描下方二维码即可一键订阅:

推荐阅读
点击“阅读原文”,追踪复杂科学顶刊论文
微信扫一扫,分享到朋友圈











