过去几十年,生物数据集的规模与复杂性大幅增长,这使得机器学习越来越多地用于为潜在生物过程构建信息与预测模型。所有机器学习技术都在让模型与数据相匹配;然而,具体的方法多种多样,乍一看似乎令人眼花缭乱。对于不同类型的生物数据,该如何选择特定的机器学习技术?
2021年9月,发表在 Nature Reviews Molecular Cell Biology 上的综述文章“给生物学家的机器学习指南”,向读者简要介绍了一些关键的机器学习技术:既包括分类、回归、聚类模型等传统机器学习方法,也包括最近开发和广泛使用的涉及深度神经网络的技术。本文还记录了一些最佳做法与入门要点,并展望了机器学习应用于生物学的一些最令人兴奋的前景。
研究领域:机器学习,人工神经网络,生物数据
Joe G. Greener, Shaun M. Kandathil等 | 作者
赵雨亭 | 译者
陈斯信 | 审校
邓一雪 | 编辑

论文题目:
A guide to machine learning for biologists
https://www.nature.com/articles/s41580-021-00407-0
人类通过观察来理解周围的世界,并学习预测接下来会发生什么。设想一个孩子学习接球:这个孩子通常对控制抛球运动的物理定律一无所知;然而,通过观察、试错的过程,她/他会调整对球运动的理解,以及如何移动身体,直到能够可靠地接住球。换句话说,这个孩子通过建立一个足够准确和有用的过程“模型”,对数据反复测试这个模型,并对模型进行修正使其更好,学会了如何接球。
“机器学习”泛指用预测模型拟合数据或识别数据中的信息分组的过程。机器学习领域本质上试图近似或模仿人类识别模式的能力——尽管是以“计算”这样客观的方式。当人们想要分析的数据集因为太大(许多单独的数据点)或太复杂(包含大量特征)而无法进行人工分析,或者当人们需要自动化数据分析的过程,来建立可重复且省时的工作流程时,机器学习就很有用。来自生物实验的数据通常就是这样的。在过去的几十年里,生物数据集的规模与复杂性都大幅增长。因此,除了掌握一些可以用来解释大量数据的实用方法,透彻理解所使用的这些技术也变得越来越重要。机器学习已经在生物领域中使用了几十年,但它的重要性稳步增长,几乎已经应用于生物学的每个领域。然而,直到过去几年中,该领域才开始对可用策略进行更严格的审视,并开始评估在不同的场景下,哪些方法最适合,或者根本不合适。
这篇综述旨在让生物学家了解如何开始理解和使用机器学习技术。本文不打算对使用机器学习解决生物问题的文章进行全面的文献综述,或描述各种机器学习方法的详细数学基础 [2, 3]。相反,本文专注于将特定技术与不同类型的生物数据联系起来(类似的综述是针对特定的生物学科的;详见参考文献 [4-11])。本文还尝试提炼出一些关于如何实际进行训练和改进模型过程的最佳做法。生物数据的复杂性为使用机器学习技术进行分析带来了陷阱和机会。为了解决这些问题,本文讨论了影响成果有效性的常见问题,并提供了如何避免这些问题的指导。这篇综述的大部分内容都致力于描述许多机器学习技术,在每种情况下,本文都提供了适当应用该方法以及如何解释结果的示例。讨论的方法包括传统的机器学习方法——它们在许多情况下仍然是最佳选择——以及基于人工神经网络的深度学习,这些方法正在成为许多任务的最有效方法。本文最后描述了将机器学习纳入生物学数据分析通用流程的未来是什么样的。
在生物学中使用机器学习有两个目标。首先是在缺乏实验数据的地方做出准确的预测,并利用这些预测来指导未来的研究工作。然而,作为科学家,我们寻求了解世界,因此第二个目标是使用机器学习来进一步了解生物学。本指南讨论了在机器学习中,这两个目标如何经常发生冲突,以及如何从通常被视为“黑箱”的模型中提取理解:毕竟它们的内部工作原理难以理解[12]。
本文首先介绍机器学习中的一些关键概念。在可能的情况下,本文会用生物学文献中的例子来说明这些概念。
一般术语。一个数据集包含许多数据点(data points)或实例(instances),每个数据点或实例都可以被认为是来自实验的单个观察。每个数据点都由一定数量(通常是固定的)的特征(features)所描述。此类特征的例子包括长度、时间、浓度和基因表达水平。机器学习的任务是对希望模型输出的值进行客观规范。例如,对于研究基因随时间表达的实验,研究人员可能想要预测特定代谢物转化为另一个物种的速率。在这种情况下,特征“基因表达水平”和“时间”可以称为输入特征或模型的简单输入,“转换率”将是模型的期望输出;也就是说,研究人员有兴趣预测的数量。一个模型可以有任意数量的输入和输出特征。特征可以是连续的(取连续数值)或分类的(只取离散值)。很多时候,分类特征只是二元的,要么是真 (1),要么是假 (0)。
监督学习和无监督学习。“监督机器学习”(supervised learning)是指将模型与已标记的数据(或数据子集)进行拟合——其中存在一些属性的真实值(ground truth),通常通过实验测量或由人类分配。例子包括对蛋白质二级结构的预测 [13] 和对基因组调控因子的基因组可及性的预测 [14]。在这两种情况下,真实值最终都来自实验室观察,但这些原始数据通常以某种方式进行了预处理。例如,在二级结构的情况下,真实值来自分析蛋白质数据库中的蛋白质晶体结构数据,在后一种情况下,真实值来自DNA测序实验的数据。相比之下,无监督学习(unsupervised learning)方法能够识别未标记数据中的模式,而无需以预定标签的形式向机器学习系统提供真实值,例如在基因表达研究中找到具有相似表达水平的患者子集[15] 或预测基因序列共变异的突变效应 [16]。有时这两种方法在半监督学习中结合,其中少量标记数据与大量未标记数据结合。在获取标记数据的成本很高的情况下,这可以提高性能。
分类、回归和聚类问题。当问题涉及将数据点分配给一组离散类别(例如,“癌性”或“非癌性”)时,该问题称为“分类问题”(classification),执行这种分类的任何算法都可以说是分类器。相比之下,回归(regression)模型输出一组连续的值,例如预测蛋白质中一个残基突变后的折叠自由能变化 [17]。连续值可以阈值化或以其他方式离散化,这意味着通常可以将回归问题重新表述为分类问题。例如,上述自由能变化可以归入对蛋白质稳定性有利或不利的值范围内。聚类(clustering)方法用于预测数据集中相似数据点的分组,通常基于数据点之间的某种相似性度量。它们是无监督方法,不需要数据集中的示例具有标签。例如,在基因表达研究中,聚类可以找到具有相似基因表达的患者子集。
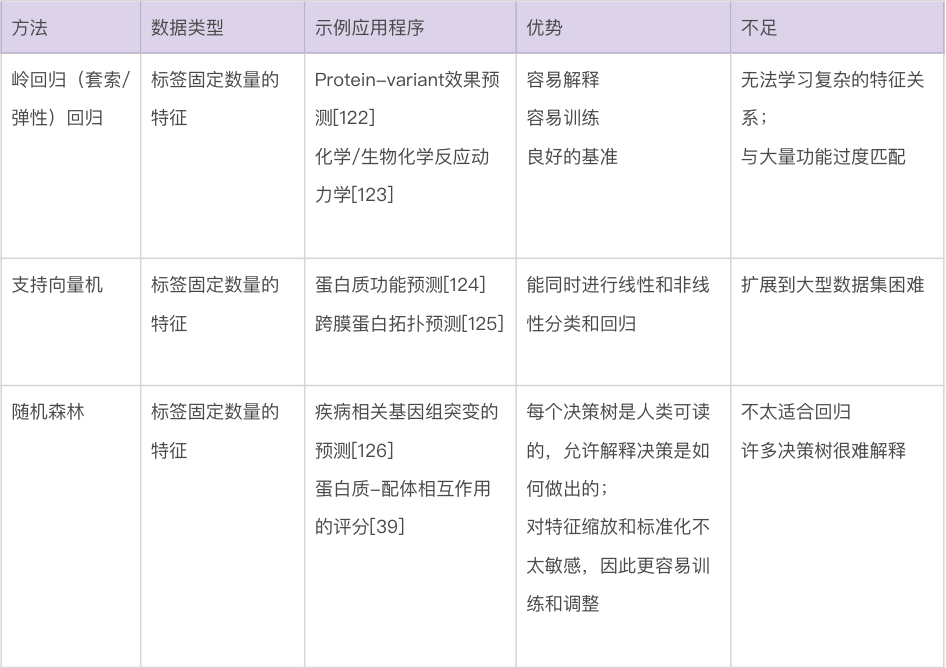
类和标签。分类器返回的离散值可以是相互排斥的,在这种情况下,它们被称为“类”(class)。当这些值不需要相互排斥时,它们被称为“标签”(label)。例如,蛋白质结构中的残基只能属于多个二级结构类别中的一个,但可以同时、不互斥地被标记为α-螺旋和跨膜。类和标签通常由编码表示(例如,独热编码,one-hot encoding)。
损失或代价函数。机器学习模型的一个或多个输出从来都不是理想的,会偏离真实值。测量这种偏差的数学函数,或更笼统地说,测量获得的输出和理想的输出之间“不一致”的程度的数学函数,被称为“损失函数”(loss function)或“代价函数”(cost function)。在监督学习设置中,损失函数将衡量模型的输出相对于真实值的偏差。例子包括回归问题的均方误差损失(mean squared error)和分类问题的二元交叉熵(binary cross entropy)。
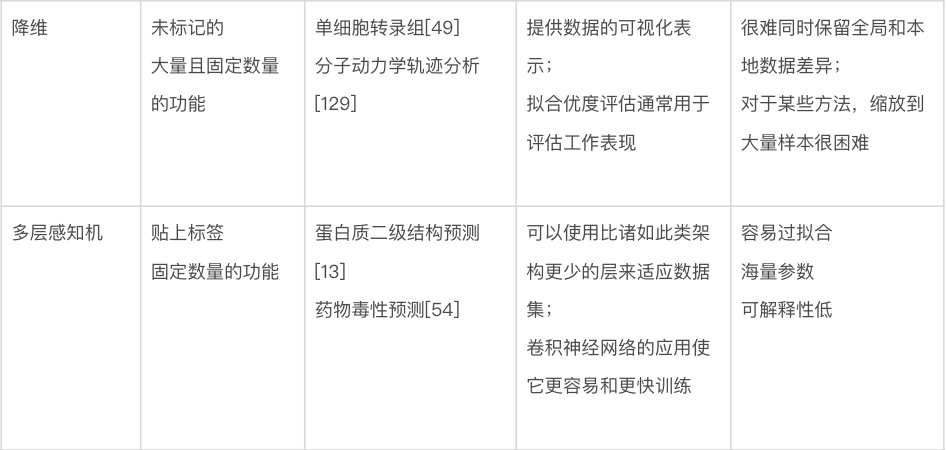
参数和超参数。模型本质上是对一组输入特征进行运算,并产生一个或多个输出值或特征的数学函数。为了能够学习训练数据,模型包含可调参数(parameter),其值可以在训练过程中改变,以实现模型的最佳性能(见下文)。例如,在一个简单的回归模型中,每个特征都有一个乘以特征值的参数,然后将它们相加以进行预测。超参数是可调整的值,不被视为模型本身的一部分,因为它们在训练期间不会更新,但仍然对模型的训练及其性能产生影响。超参数的一个常见示例是学习率(learning rate),它控制模型参数在训练期间更改的速率或速度。
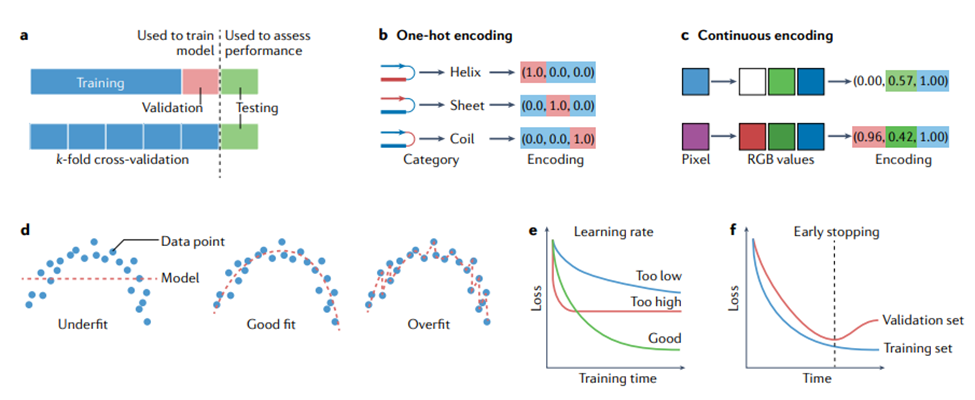
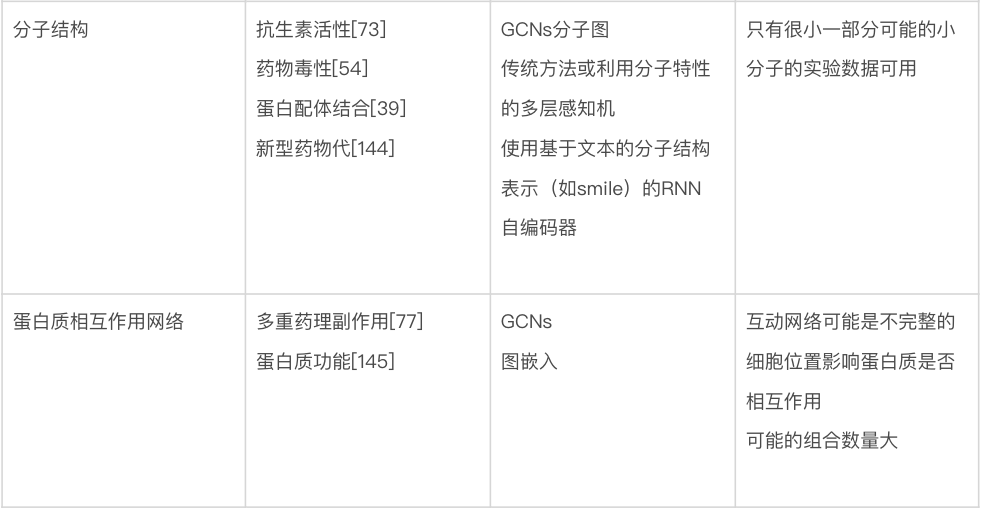
训练、验证和测试。在用于进行预测之前,模型需要进行训练(training),这涉及自动调整模型的参数以提高其性能。在监督学习设置中,这涉及通过最小化损失或代价函数的平均值(前面已描述)来修改参数,使模型在训练数据集上表现良好。通常,单独的验证(validation)数据集用于监控但不影响训练过程,以检测潜在的过度拟合(参见下一节)。在无监督设置中,代价函数仍然被最小化,尽管它不再涉及真实值。一旦模型经过训练,就可以在未用于训练的数据上进行测试(testing)。有关整个训练过程以及如何在训练集和测试集之间适当拆分数据的指南,请参见方框1。图1展示了整个机器学习过程的流程图。图2展示了模型训练中的一些概念。
过拟合和欠拟合。将模型拟合到训练数据的目的是捕捉数据中变量之间的“真实”关系,以便模型对本次未包含的(即非训练)数据具有预测能力。过拟合或欠拟合的模型将对不在训练集中的数据产生较差的预测(图 2d)。过拟合的模型会在训练集中的数据上产生很好的结果(通常是因为参数太多),但在真实数据上会产生很差的结果。图2d中的过拟合模型正好通过每个训练点,因此它在训练集上的预测误差为零。然而,很明显,这个模型已经“记住”了训练数据,不太可能在真实数据上产生好的结果。相比之下,欠拟合的模型无法充分捕捉数据中变量之间的关系。这可能是由于模型类型选择不正确、对数据的假设不完整或不正确、模型中的参数太少和/或训练过程不完整。如图2d 所示的欠拟合模型对于它试图拟合的数据来说是不够的;在这种情况下,很明显变量具有非线性关系,不能用简单的线性模型充分描述。因此,非线性模型更合适。
归纳偏差和偏差-方差权衡。模型的“归纳偏差”是指在学习算法中做出的一组假设,这些假设导致它偏向于学习问题的特定解决方案,而不是其他解决方案。它可以被认为是模型对学习问题的特定类型解决方案的偏好。这种偏好通常使用其特定的数学形式和/或使用特定的损失函数被编程到模型中。例如,循环神经网络(recurrent neural networks,RNN;下文中将进行讲解)的归纳偏差是输入数据中存在顺序依赖性,例如代谢物随时间的浓度。这种依赖关系在 RNN 的数学形式中得到了明确的解释。不同模型类型中的不同归纳偏差使它们更适合并且通常对特定类型的数据表现更好。另一个重要概念是偏差(bias)和方差(variance)之间的权衡。可以说具有高偏差的模型对训练模型具有更强的约束,而具有低偏差的模型对被建模的属性做出较少的假设,并且理论上可以对多种函数类型进行建模。模型的方差描述了在不同训练数据集上进行训练,经过训练的模型发生的变化的大小。一般来说,研究人员希望模型具有非常低的偏差和低方差,尽管这些目标经常发生冲突,因为具有低偏差的模型通常会在不同的训练集上学习不同的信号。控制偏差-方差权衡是避免过拟合或欠拟合的关键。
方框1 | 做机器学习
这个方框概述了在训练机器学习模型时应该采取的步骤。令人惊讶的是,关于模型选择和训练过程的指导很少 [146,147],对垫脚石和失败模型的描述也很少被纳入已发表的研究文章。在接触任何机器学习代码之前,第一步应该是充分理解手头的数据(输入)和预测任务(输出)。这意味着对问题的生物学理解:例如了解数据的来源和噪声源,并知道,根据生物学原理,输出理论上是怎样从输入中预测的。例如,可以推断不同的氨基酸可能对蛋白质中的特定二级结构有偏好,因此,从蛋白质序列中每个位置的氨基酸频率预测二级结构是有意义的。了解输入和输出在计算上是如何存储的也很重要。它们是否已被归一化以防止一个特征对预测产生过大的影响?它们是编码为二进制变量还是连续编码?是否有重复的条目?是否缺少数据元素?
接下来,应拆分数据以进行训练、验证和测试。有多种方法可以做到这一点,图 2 中显示了其中的两种方法。2a. 训练集用于直接更新被训练模型的参数。验证集通常占可用数据的 10% 左右,用于监控训练、选择超参数并防止模型过度拟合训练数据。通常使用k折交叉验证(k-fold cross validation):将训练集分成 k 个大小均匀的分区(例如,五个或十个)以形成 k 个不同的训练和验证集,并在每个分区之间比较性能以选择最佳超参数。测试集,有时也称为“留出集”,通常也占可用数据的 10% 左右,用于评估模型在未用于训练或验证的数据上的性能(即估计其预期的真实表现)。测试集应该只在研究的最后使用一次,或者尽可能不频繁地使用 [27, 38] ,以避免模型拟合了测试集。关于制作一个公平的测试集时需要考虑的问题,请参见数据泄漏一节。
下一步是模型选择,这取决于数据的性质和预测任务,总结在图1中。1. 训练集用于训练模型,按照所用软件框架的最佳做法。为达到最佳性能,大多数方法都有一些超参数需要调整。这可以使用随机搜索或网格搜索来完成,并且可以与上面概述的k折交叉验证相结合 [27]。研究人员应该考虑模型集成,将多个相似模型的输出简单平均,以提供一种相对可靠的方法,来提高建模任务的整体准确性。最后,应该评估模型在测试集(见上文)上的准确性。
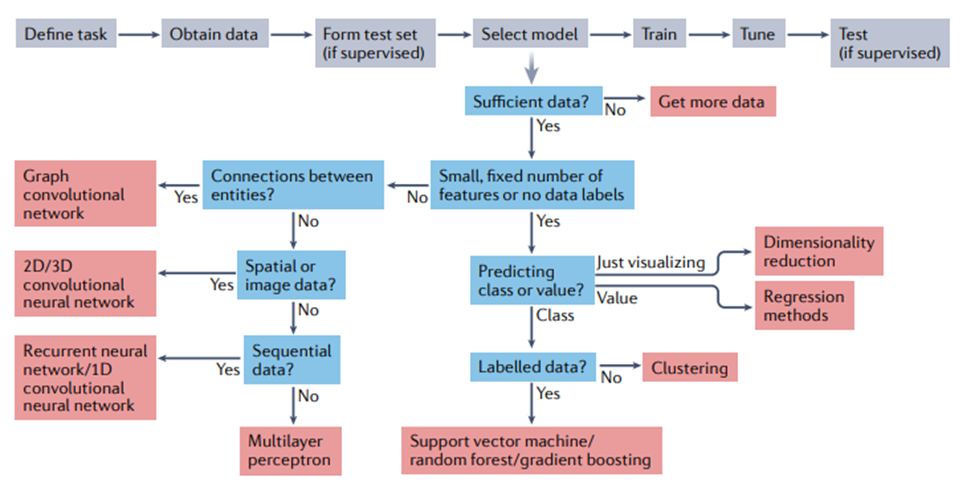
图1. 选择和训练机器学习方法。训练机器学习方法的整个过程显示在上部。下面给出了帮助研究人员选择模型的决策树。这个流程图旨在作为一个视觉指南,将本综述中概述的概念联系起来。然而,像这样的简单概述不能涵盖所有情况。例如,机器学习要变得适用所需的数据点数量,取决于每个数据点可用的特征数量——特征越多,需要的数据点越多,还取决于所使用的模型。还有一些深度学习模型可以处理未标记的数据。
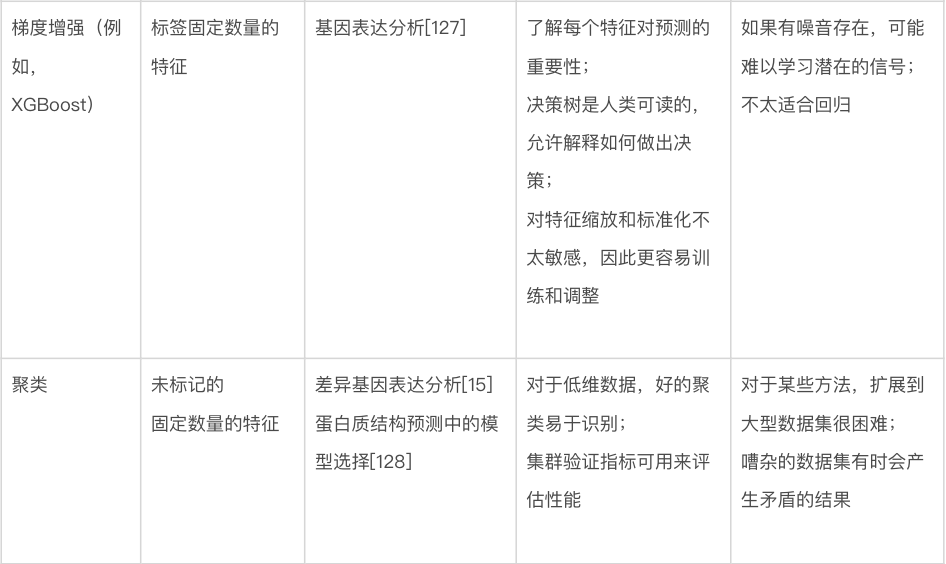
图2. 训练机器学习方法。(a)可用数据通常分为训练、验证和测试集。训练集直接用于训练模型,验证集用于监控训练,测试集用于评估模型的性能。也可以使用带有测试集的k折交叉验证。(b) 独热编码是表示分类输入的常用方法,只允许从多种可能性(在这里是三种可能的蛋白质二级结构类别)中进行单一选择。编码的结果是一个包含三个数字的向量,除了被占用的类设置为 1 外,所有数字都等于 0。这个向量被机器学习模型使用。(c)连续编码表示数字输入,在这种情况下是图像中像素的红色、绿色和蓝色 (RGB) 值。结果同样是一个包含三个数字的向量,对应于像素中红色、绿色和蓝色的数量。(d)未能学习到变量之间的潜在关系称为“欠拟合”,而学习了训练数据中的噪声称为“过拟合”。欠拟合可能是由于使用了不够复杂的模型来描述信号。而过拟合可能是由于使用了参数过多的模型或在学习了变量之间的真实关系后继续训练。(e)模型的学习率决定了在训练神经网络或一些传统模型(如梯度提升)时调整学习参数的速度。低学习率会导致训练缓慢,这既耗时又需要相当大的计算能力。相反,高学习率会导致快速收敛到一个非最优解和模型性能不佳。(f)提前停止(early stopping)是在验证集上的损失函数开始增加时终止训练的过程,即使训练集上的损失函数仍在减少。使用提前停止可以防止过拟合。
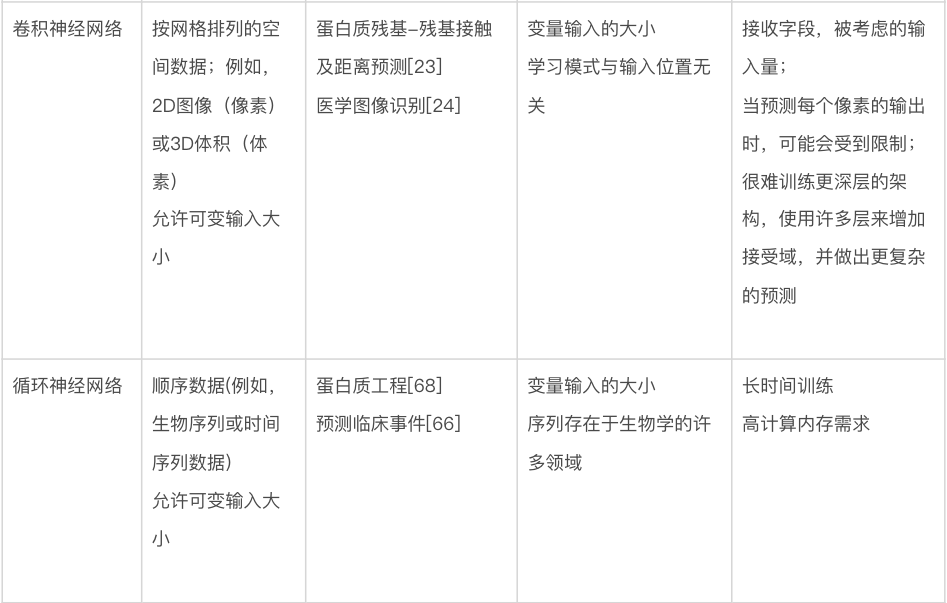
本文将讨论几种关键的机器学习方法,重点介绍它们的特定优势和劣势。表1显示了不同机器学习方法的比较。本节将首先讨论不基于神经网络的方法,有时称为“传统机器学习”。图 3 显示了传统机器学习的一些方法。此类模型可以使用各种软件包来训练,包括 Python [18] 中的 scikit-learn、R [19] 中的 caret 和 Julia [20] 中的 MLJ。
当开发用于生物数据的机器学习方法,为给定任务寻找最合适方法时,通常应将传统机器学习视为第一个探索领域。深度学习的确是一种强大且目前无疑流行的工具。但是,深度学习在其擅长的应用领域仍然有三个要求:有大量可用数据(例如,数百万个数据点)、每个数据点有很多特征、特征高度结构化(特征彼此之间有明确的关系,例如图像中的相邻像素)[21]。生物数据可以满足这些要求,并且深度学习已成功应用的生物数据的例子包括 DNA、RNA 和蛋白质序列 [22, 23] 以及显微镜图像 [24, 25] 等数据。然而,即使满足其它两个要求,对大量数据的要求也可能使深度学习成为一个糟糕的选择。
与传统方法相比,深度学习开发和测试给定问题的速度要慢得多。与支持向量机 (SVM) 和随机森林 [27] 等传统模型相比,开发深度神经网络的架构然后对其进行训练可能是一项耗时且计算成本高的任务 [26]。尽管存在一些方法,但对于深度神经网络,估计特征重要性 [28](即每个特征对预测的重要性)或模型预测的置信度仍然不是一件容易的事 [1, 28, 29 ],这两者在生物环境中通常是必不可少的。即使对于特定的,深度学习在技术上似乎可行的生物预测任务,谨慎的做法通常还是:训练传统方法(如果可能的话),再将其与基于神经网络的模型进行比较[30]。
传统方法通常期望数据集中的每个示例具有相同数量的特征,因此这并不总是可行的。一个明显的生物学示例是,在使用蛋白质、RNA 或 DNA 序列的时候,每个示例具有不同的长度。要对这些数据使用传统方法,可以使用简单的技术(例如填充和加窗)更改数据,使它们的大小都相同。“填充”(padding)意味着对每个示例添加包含零的附加值,直到它与数据集中最大的示例大小相同。相比之下,加窗(windowing)将单个示例缩短到给定的大小(例如,在一个序列长度至少为100的蛋白质序列数据集中,只使用每个蛋白质的前 100 个残基)。
使用分类和回归模型。对于如图3a所示的回归问题,岭回归(具有正则化项的线性回归)通常是开发模型的良好起点,因为它可以为给定任务提供快速且易于理解的基准。当希望模型依赖可用数据中的最少特征时,线性回归的其他变体(例如 LASSO 回归 [31] 和弹性网络回归 [32])也值得考虑。不幸的是,数据中特征之间的关系通常是非线性的。对于这些情况,使用诸如SVM(support vector machine,支持向量机)之类的模型通常是更合适的选择 [33]。SVM 是一种强大的回归和分类模型,它使用核函数(kernel function)将不可分的问题转换为更容易解决的可分问题。SVM 可用于执行线性回归和非线性回归,具体取决于使用的核函数 [34-37] 。开发模型的一个好方法是训练一个线性 SVM 和一个带有径向基函数核的 SVM(一种通用非线性类型的 SVM)来量化可以从非线性函数中获得的增益(如果有的话)。非线性方法可以提供更强大的模型,但代价是不容易解释哪些特征影响模型。这就是前面所提到的偏差-方差权衡。
回归中常用的许多模型也用于分类。训练线性 SVM 和带有径向基函数核的 SVM 也是分类任务的一个很好的默认起点。另一种可以尝试的方法是k 最近邻(k nearest neighbours)分类[38]。作为最简单的分类方法之一,k 最近邻分类提供了一个有用的基线性能标记,可以与其他更复杂的模型(例如 SVM)进行比较。另一类稳健的非线性方法是基于集成(ensemble)的模型,例如随机森林 [39] 和 XGBoost [40, 41]。这两种方法都是强大的非线性模型,好处在于能额外提供特征重要性的估计,并且通常需要最少的超参数调整。如果某个生物学任务需要了解哪些特征对预测的贡献最大才能理解,那么这些模型就是一个不错的选择,毕竟它们能分配特征重要性值,有决策树结构,
对于分类和回归,许多可用的模型往往具有令人眼花缭乱的特点和变体。试图先验地预测特定方法对特定问题的适用程度可能靠不住,而采用经验、反复试验的方法来寻找最佳模型通常是最谨慎的方法。使用现代机器学习函数——例如 scikit-learn [18]——在这些模型变体之间进行更改,通常只需要更改一行代码。因此,选择最佳方法的一个好的总体策略是:训练和优化上述各种方法,选择在验证集上性能最好的那个,最终比较它们在单独的测试集上的性能。
使用聚类模型。聚类算法(图 3e)的使用在生物学中很普遍 [42、43]。k平均(k-means)是一种强大的通用聚类方法,与许多其他聚类算法一样,它需要将聚类数设置为超参数 [44]。DBSCAN 是一种替代方法,它不需要预定义集群的数量,但代价是需要设置其他超参数 [45] 。也可以在聚类之前执行降维,以提高在具有大量特征的数据集上的性能。
降维。降维(dimensionality reduction)技术用于将具有大量属性(或维度)的数据转换为低维形式,同时尽可能保留数据点之间的不同关系。例如,相似的数据点(例如,两个同源蛋白质序列)在其低维形式上也应该相似,而不同的数据点(例如,不相关的蛋白质序列)应该保持不相似[46,47]。通常选择两个或三个维度来允许在一组轴上可视化数据,尽管更多的维度也用于机器学习。这些技术包括数据的线性和非线性变换。生物学中常用的例子包括主成分分析(principle component analysis,PCA,如图3)、UMAP(Uniform Manifold Approximation and Projection)和t-SNE(t-distributed stochastic neighbour embedding)。生物学数据处理中具体使用的技术取决于情况:PCA 保留数据点之间的全局关系并且是可解释的,因为每个组件都是一个输入特征的线性组合,这意味着很容易理解哪些特征会导致数据的多样性。t-SNE 更好地保留了数据点之间的局部关系,是一种灵活的方法,可以揭示复杂数据集中的结构。应用包括用于 t-SNE[49] 的单细胞转录组学和用于主成分分析的分子动力学轨迹分析。
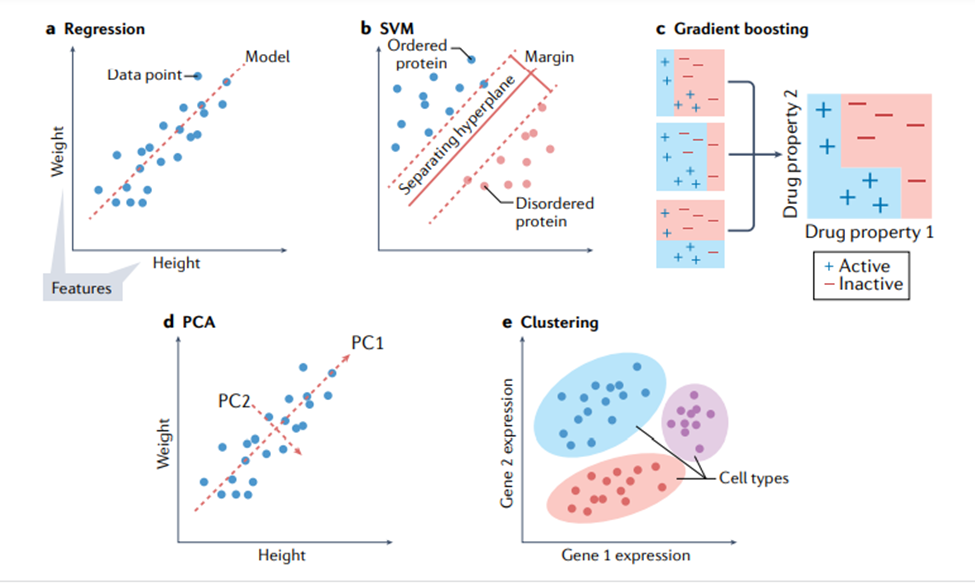
图3. 传统的机器学习方法。(a) 回归寻找因变量(观察到的属性)和一个或多个自变量(特征)之间的关系;例如,从一个人的身高预测一个人的体重。(b) 支持向量机 (SVM) 转换原始输入数据,以便在它们的转换版本(称为“潜在表示”)中,属于不同类别的数据被尽可能宽的明显间隙划分。这里展示了对蛋白质是有序还是无序的预测,横轴与纵轴代表转换数据的维度。(c) 梯度提升使用一组弱预测模型(通常是决策树)来进行预测。例如,活性药物可以从分子描述符(例如分子量和特定化学基团的存在)中预测。各个预测器以阶段方式组合以进行最终预测。(d) 主成分分析 (PCA) 会发现一系列特征组合,这些特征组合在彼此正交的情况下最能描述数据。它通常用于降维。在“身高与体重”的例子中,对应于身高和体重的线性组合的第一主成分 (first principal component,PC1) 描述了强正相关,而 PC2 可能描述了与身高与体重不强相关的其他变量,例如体脂百分比或肌肉质量百分比。(e) 聚类使用各种算法中的一种,来对相似的对象进行分组(例如,根据基因表达谱对细胞类型进行分组)。
人工神经网络(artifical neural networks)模型会有这个名字,是因为要拟合的数学模型的形式受到大脑中神经元的连通性和行为的启发,以及这个模型最初旨在了解大脑功能 [50]。然而,数据科学中常用的神经网络作为大脑模型已经过时,现在,它只是机器学习模型,可以在某些应用中提供最先进性能。由于深度神经网络的架构和训练的快速发展,近几十年来,人们对神经网络模型的兴趣不断增长 [26]。本节将描述基本的神经网络,以及广泛用于生物学研究的变体。其中一些如图4所示。
神经网络的基本原理。神经网络的一个关键特性是它们是通用函数逼近器,这意味着,只需很少的假设,正确配置的神经网络就可以将任何数学函数逼近任意精度水平。换句话说,如果任何过程(生物的或其他的)可以被认为是一组变量的某种函数,那么该过程可以被建模到任意程度的准确度,仅受模型的大小或复杂性的控制。上述对通用逼近的定义在数学上并不严谨,但确实突出了对神经网络的兴趣持续数十年的一个原因。然而,这种保证并没有提供一种找到神经网络模型的最佳参数,为给定的数据集产生最佳近似值的方法,也不能保证该模型将为新数据提供准确的预测 [51]。
人工神经元是所有神经网络模型的基石。一个人工神经元只是一个数学函数,以特定方式将输入映射(转换)为输出。单个人工神经元接受任意数量的输入值,对它们应用特定的数学函数并返回输出值。使用的函数通常表示为:
其中xi 表示单个输入变量或特征(有 n 个这样的输入),wi 表示该输入的可学习权重(weight),b 表示可学习的偏差项,σ 表示接受单个输入并返回单个输入的非线性激活函数输出。创建网络的过程中,人工神经元按层排列,一层的输出作为下一层的输入。网络的节点可以被认为是持有上述方程中的 y 值,这些值成为下一层的 x 值。以下小节描述了排列人工神经元的各种方法,这些小节称为“神经网络架构”。结合不同的架构类型也很常见;例如,在用于分类的卷积神经网络 (CNN) 中,通常使用全连接层来产生最终的分类输出。
多层感知机(multilayer perceptron)。神经网络模型最基本的布局是以全连接方式排列的人工神经元层,如图4a所示。在这个布局中,固定数量的“输入神经元”代表根据输入网络的数据计算的输入特征值,一对神经元之间的每个连接代表一个可训练的权重参数。这些权重是神经网络中主要的可调参数,优化这些权重就是神经网络训练的意义所在。在网络的另一端,许多输出神经元代表网络的最终输出值。这种网络在正确配置后可用于对输入做出复杂的分层决策,因为给定层中的每个神经元都接收来自前一层中所有神经元的输入。这种简单排列的神经元层通常被称为“多层感知机”,是第一个可用于生物信息学应用的网络 [52, 53]。由于其训练的简便性和速度,它们今天仍广泛用于许多生物建模应用程序 [13, 54]。然而,在许多其他应用中,这些简单的架构已经被下面讨论的较新的模型架构所超越,尽管其中一些较新的架构仍然经常使用全连接层作为子组件。
卷积神经网络(CNN)。CNN 非常适合图像类数据,当数据具有某种类型的局部结构,并且识别这种结构是分析的关键目标的时候。以图像为例,这种局部结构可能与视野中特定类型的对象(例如,显微镜图像中的细胞)相关,在输入图像中由特定局部颜色模式和/或空间接近像素中的边缘表示。
CNN 由一个或多个卷积层组成(见图 4b),其中输出是将一个小的、一层全连接的神经网络(称为“过滤器”(filter)或“核”(kernel))应用于输入中的局部特征的结果。如果输入是类似图像的,该局部区域就是图像中的一小块像素。卷积层的输出也是类似图像的数组,包含滤波器在整个输入上“滑动”并在每个位置计算输出的结果。至关重要的是,所有像素都使用相同的过滤器,允许过滤器学习输入数据中的局部结构。在更深的 CNN 中,使用跳跃连接(skip connection)是很常见的,除了通过层中的处理单元之外,还允许输入信号绕过一个或多个层。这种类型的网络称为“残差网络”(residual network),可以让训练更快地收敛到准确的解决方案上。
CNN 经过配置,对不同空间结构的数据都可以进行有效操作。例如,一维 CNN 的过滤器只会在一个方向上滑动(比如从左到右);这种类型的 CNN 适用于只有一个空间维度的数据(例如文本或生物序列)。二维 CNN 对具有两个空间维度的数据进行操作,例如数字图像。三维 CNN 对体积数据进行操作,例如磁共振成像扫描。
在生物学中,CNN 针对各种数据类型都已取得了重大成功。蛋白质结构预测的最新进展是使用相关蛋白质序列中残基对共同进化的信息,来提取残基对接触和距离的信息,从而能够以前所未有的准确度建立对三维蛋白质结构的预测 [23,55]。在这种情况下,网络学习挑选出直接耦合的相互作用,即使一个序列只有很少或没有相关序列,也可以对它做出准确的预测 [56]。CNN 也已成功应用于识别基因序列数据中的变异 [57] 、三维基因组折叠 [58] 、DNA-蛋白质相互作用 [22, 59] 、低温电子显微镜图像分析 [60, 61] 和医学重要领域的图像分类。目前,它们在例如检测恶性肿瘤之类的领域,已经可以与人类专家的表现相媲美[24,62]。
循环神经网络(RNN)。RNN 最适合于有序序列(ordered sequences)形式的数据,在这些序列中,一个点和下一个点之间(至少在理论上)存在一些相关性或相关性。它们在生物学之外的应用,可能主要是在自然语言处理中,其中文本被视为一系列单词或字符。如图 4c 所示,RNN 可以被认为是一个神经网络层块,它将序列中每个条目(或时间步长)对应的数据作为输入,并为每个条目生成一个输出,该输出依赖于先前处理过的条目。它们还可用于生成整个序列的表示,该表示传递到网络的后续层以生成输出。这在科研中优势巨大:因为任何长度的序列都可以转换为固定大小的表示并输入到多层感知机。在生物学中使用 RNN 效果最明显的例子是分析基因或蛋白质序列:其任务包括从基因序列中识别启动子区域、预测蛋白质二级结构,以及建模基因表达水平随时间的变化;在最后一种情况下,给定时间点的值将计为序列中的一个条目。RNN 的更先进的变体,长短期记忆(long short-term memory)或门控循环单元(gated recurrent unit),在生物学中有许多用途,包括蛋白质结构预测 [63, 64] 、肽设计 [65] 和根据健康记录预测临床诊断 [66] 。这些更高级的方法通常与 CNN 结合使用,可以提高准确性 [67]。RNN 在分析基于序列的数据时非常稳健。例如,在数百万个蛋白质序列上训练的 RNN 已经显示出捕捉进化和结构信息的能力,并且可以应用于各种监督任务,包括与新蛋白质序列设计相关的任务 [68]。
注意力机制的作用和Transformer的使用。RNN 发现的一个问题是它们在检查输入序列的特定部分(对于生成高度准确的输出很重要)时遇到困难。向 RNN 添加注意力机制(attention mechanism),允许模型在计算每个输出时访问输入序列的所有部分,以缓解这个问题。最近的研究表明,甚至根本不需要 RNN ,单独使用注意力机制就可以了——由此产生的模型被称为“转换器”(transformer),在许多自然语言处理基准测试中获得了优秀的预测结果[69]。Transformer 模型最近在生物序列任务上表现出比 RNN 更高的准确性,但这些方法(通常使用数千个图形处理单元(GPU)对数十亿个序列进行训练)是否能够胜过现有的基于对齐的方式,还有待观察。基于生物信息学中序列分析的方法[70]。在14 届蛋白质结构预测关键评估(CASP14)实验中,AlphaFold2 取得了巨大成功。由于这个对从序列预测蛋白质结构的计算方法的评估是盲的,该结果表明使用注意力的模型也有望用于结构生物学中的任务 [71] 。
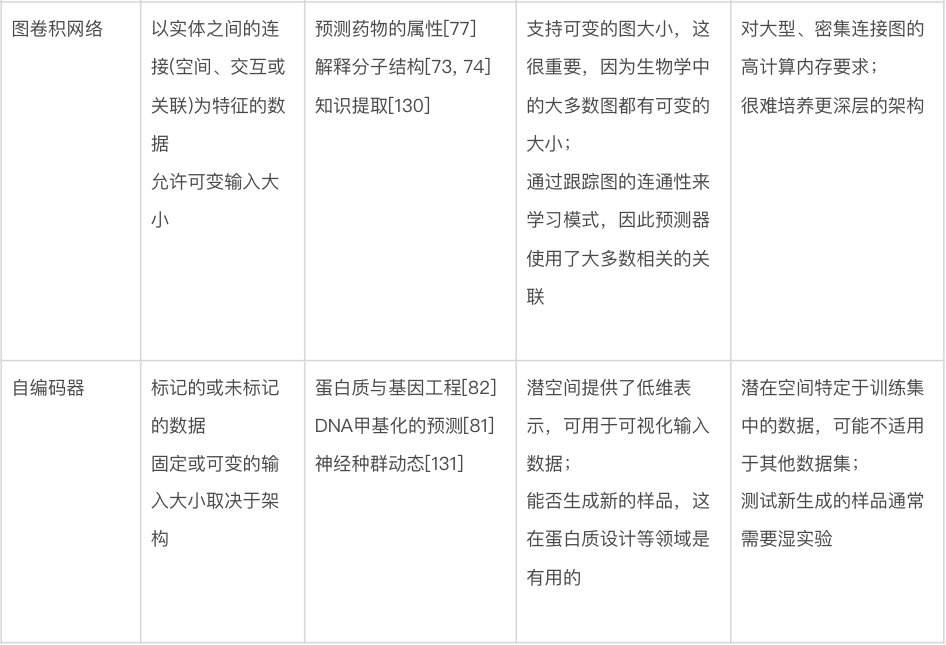
图卷积网络。图卷积网络(graph convolutional networks)特别适用这类数据——虽然没有任何明显的可见结构(如图像),但仍然由通过任意指定关系或交互连接的实体组成 [72]。与生物学相关的此类数据的例子包括分子(由原子和键组成)[73-76] 和蛋白质-蛋白质相互作用网络(由蛋白质和相互作用组成)[77]。在计算术语中,图(graph)只是此类数据的表示,每个图都有一组顶点(vertex)或节点(node),以及一组表示节点之间各种类型关系或连接的边(edge)。用上面给出的例子来解释的话,原子或蛋白质的表示可能被归类为节点特征(node feature),而键或相互作用可能被归类为边特征(edge feature)。图卷积网络使用以此产生的图结构来确定神经网络模型中的信息流。如图4d所示,当每个节点在整个网络中更新特征时,会考虑相邻节点(adjacent node),最后一层的节点特征被用作输出(例如,蛋白质上的相互作用残基)或组合形成整个输出图(例如,蛋白质的折叠类型)。代表着不同关联的图可以在进行预测时结合不同的信息来源,例如结合药物-基因和食物-基因关系图来预测预防癌症的食物[78]。用于训练图卷积网络的软件包括 PyTorch Geometric[79] 和 Graph Nets[72]。
自编码器(autoencoder)。顾名思义,自编码器是一种神经网络架构,通过将数据点表示为在具有预定维度(通常远小于输入维度)的新空间中的点,来对数据点集合进行自编码(autoencode)。训练一个神经网络(编码器,encoder)将输入转换为紧凑的内部表示,称为“潜在向量”(latent vector)或“潜在表示”(latent representation),表示新空间中的单个点。自编码器的第二部分称为“解码器”(decoder),将潜在向量作为输入,经过训练产生具有原始维度的原始数据,作为输出(图 4e)。另一种看待这个问题的方法是编码器尝试压缩输入,而解码器尝试解压缩。编码器、潜在表示和解码器同时训练。尽管这种“输出模仿输入”的做法听起来似乎毫无意义,但其目的是学习输入数据的新表示,使得该表示能紧凑地编码所需的特征,例如数据点之间的相似性,同时仍然保留重建原始数据的能力。应用包括预测两个数据点的关联程度,以及强行让数据在潜在空间上形成一些对进一步预测任务有用的结构。编码器-解码器架构的另一个好处是,一旦经过训练,就可以单独使用解码器来生成(预测)新的合成数据样本。这些样本可以在实验室中进行测试,有助于合成生物学的研究 [80] 。自编码器已应用于一系列生物学问题,包括预测 DNA 甲基化状态 [81]、基因和蛋白质序列工程 [82、83] 以及单细胞 RNA 测序分析 [84]。
训练和改进神经网络。方框1 概述了训练机器学习模型的一般程序。但是,由于神经网络在结构上比传统机器学习算法复杂得多,因此存在一些神经网络特有的问题。在选择了一个神经网络作为适合预期应用的模型(图1)后,先只在单个训练示例(例如,单个图像或基因序列)上对其进行训练,通常是个好主意。这种经过训练的模型对于进行预测没有用处,但训练可以很好地揭示编程错误。训练损失函数应该很快变为零,因为网络只是简单地记住输入;如果不是,则代码中可能存在错误,或者算法不够复杂,无法对输入数据进行建模。一旦网络通过了这个基本的调试测试,就可以继续对整个训练集进行训练,来最小化训练损失函数。这可能需要调整学习率等超参数(图 2e)。通过监控训练集和验证集的损失,可以检测到网络的过度拟合——训练损失继续降低,验证集的损失开始增加。训练通常在那个时候停止,这个过程被称为提前停止(图 2f)。神经网络(或任何机器学习模型)的过度拟合,如图 1d所示,意味着模型开始简单地记住训练集的特征,因此开始失去泛化到新数据的能力。提前停止是防止这种情况的好方法,但在训练期间可以使用其他技术,例如模型的正则化(regularization)或丢弃(dropout)技术。丢弃技术随机忽略网络中的一些节点,来迫使网络学习涉及多个节点的更强大的预测策略。
用于训练神经网络的流行软件包包括 PyTorch [85] 和 Tensorflow [86]。训练神经网络的计算要求很高,通常需要具有足够内存的图形处理单元或张量处理单元,因为这些设备可以提供比使用标准中央处理单元 10 到 100 倍的加速。在训练近年来取得成功的大型模型以及在大型数据集上进行训练时,需要这种加速。然而,运行一个已经训练好的模型通常要快得多,而且通常只在普通的中央处理单元上是可行的。那些无法访问图形处理单元进行训练的人,也可以使用通用提供商的云计算解决方案。值得注意的是,对于小任务,Colaboratory (Colab) 允许在图形处理单元或张量处理单元上免费测试 Python 代码。使用 Colab 是开始使用基于 Python 的深度学习的绝佳方式。
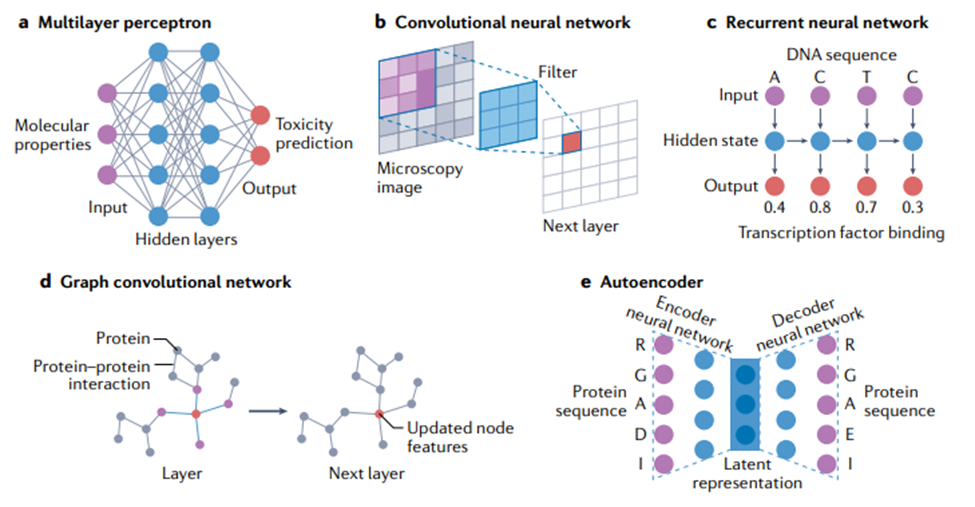
图4. 神经网络方法。(a) 多层感知机由节点(显示为圆)组成,代表数字:输入值、输出值或内部(隐藏)值。节点排列在具有连接的层中,这些连接意味着学习后的参数,位于一层的每个节点和下一层的每个节点之间。例如,分子特性可用于预测药物毒性,因为预测可以从独立输入特征(分子特性)的一些复杂组合中进行。(b) 卷积神经网络 (CNN) 使用跨输入层移动的过滤器,来计算下一层中的值。过滤器跨整个层运行,意味着参数是共享的,无论位置如何,都可以检测到相似的实体。二维 CNN 显示在显微镜图像上运行,但一维和三维 CNN 在生物学中也有应用。这里的维度指的是数据的空间维度;与此相对应,可以配置 CNN 内部的连接性。例如,生物序列可以被认为是一维的,而磁共振成像数据可以被认为是三维的。(c) 循环神经网络 (RNN) 使用相同的学到的参数来处理顺序输入的每个部分,为每个输入提供输出和更新的隐藏状态。隐藏状态用于携带序列前面部分的信息。在这个例子里,可以预测 DNA 序列中每个碱基与转录因子的结合概率。RNN 被展开,方便展示每个输出是如何使用相同的层生成的;不应该被混淆为是使用不同的层。(d) 图卷积网络使用来自图中连接节点的信息(例如蛋白质-蛋白质相互作用网络),通过组合来自所有相邻节点的预测,来更新网络中的节点属性。更新后的节点属性形成网络中的下一层,并在输出层中预测所需的属性。(e) 自编码器由编码器神经网络和解码器神经网络组成,编码器神经网络将输入转换为低维潜在表示,解码器神经网络将这种潜在表示转换回原始输入形式。例如,可以编码蛋白质序列并使用潜在表示来生成新的蛋白质序列。在示例中,5 个残基中有 4 个与自动编码器编码和解码后的输入相同,表明在该序列上的准确率为 80%。
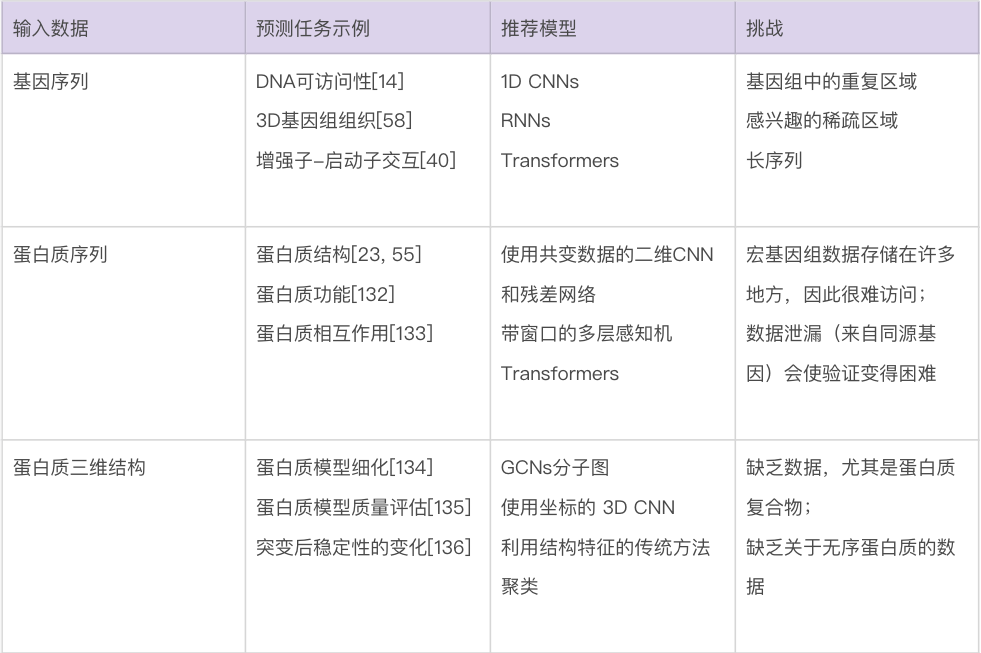
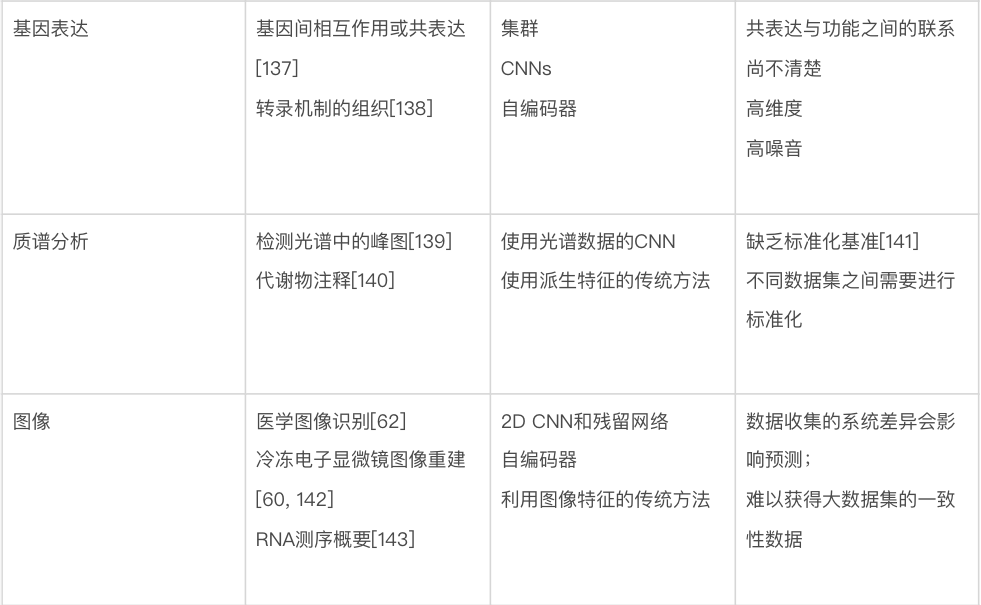
生物数据建模的最大挑战也许是种类繁多。生物学家使用的数据包括基因和蛋白质序列、基因随时间的表达水平、进化树、显微镜图像、3D 结构和相互作用网络等。表2 总结了针对特定生物数据类型的一些最佳做法和重要考虑因素。由于遇到的数据类型的多样性,生物数据通常需要一些定制的解决方案来有效地处理。要在这些问题领域中使用机器学习,很难直接推荐已有模型,更不要说通用指南,因为模型、训练程序和测试数据的选择将在很大程度上取决于人们想要回答的确切问题。尽管如此,要成功使用机器学习(不管是在生物学中还是在更广泛的意义上),还是有一些常见的问题需要考虑。
数据可得性。生物学的独特之处在于,有一些问题领域的公开数据量非常大,而其它问题领域的数据量非常小。比如,公共数据库(如 GenBank 和 UniProt)中的生物序列数据相对丰富,而关于蛋白质相互作用的可靠数据则很难获得。可用于给定问题的数据量,对可以有效使用的技术的选择,具有深远的影响。一个非常粗略的指导方针是,当只有少量数据(如三四位数)可用时,人们基本上被迫使用更传统的机器学习方法——更有可能产生可靠的预测。当有更多的数量可用时,研究人员可以开始考虑更多参数化的模型——例如深度神经网络。在有监督的机器学习中,还应该考虑数据集中每个真实标签的相对比例,如果某些标签很少见,机器学习需要更多的数据[87]。
数据泄露。尽管生物数据的规模和复杂性可能使它们看起来非常适合机器学习,但仍有一些重要的考虑因素需要牢记 [21, 88, 89]。一个关键问题是如何验证模型的性能。训练集、验证集和测试集的共同设置可能会导致一些问题,例如研究人员使用各种模型,在同一个测试集上重复测试,以获得最大的准确度,从而有高估其性能的风险,无法将其推广到其他测试集或新的测试集。然而,生物数据提出了一个更重要的问题:在具有相关条目的大型数据集(例如,由于家族关系或进化关系)中,如何确保两个密切相关的条目最终不会被分到训练集和测试集里去?如果发生这种情况,则研究人员测试的,是模型记住特定案例的能力,而不是其预测相关属性的能力。这是通常被称为“数据泄漏”(Data Leakage)的问题的一个例子,并导致结果看起来比实际情况好,这可能是研究人员不愿意严格对待这个问题的原因质疑。其它类型的数据泄漏也是可能的(例如,在训练期间使用任何在测试期间不可用的数据或特征)。接下来的讨论,主要关注于训练集和测试集中样本是否相关。
这里所说的“相关”是什么意思,取决于研究的性质。可能是从同一患者或同一生物体采样数据的情况。然而,生物学中发生数据泄漏的经典情况,出现在对蛋白质序列和结构的研究中。比较常见但通常错误的是,研究人员试图确保训练集中的蛋白质与测试集中的任何蛋白质的序列同一性都不会超过某个阈值,通常阈值为 30% 或 25%。这足以排除许多同源蛋白质对,但人们已经知道几十年了的是,一些同源蛋白质几乎没有序列相似性 [90, 91],这意味着简单地按序列同一性过滤,不足以防止数据泄漏。这对于将序列比对或序列配置文件作为输入进行操作的模型尤为重要,因为尽管两个单独的蛋白质序列可能没有任何明显的相似性,但它们的生物学功能实际上可能相同。这意味着对于机器学习模型,这两个标签本质上是相同的数据点——都在描述相同的蛋白质家族。对于蛋白质序列,避免此问题的一种解决方案是使用敏感的隐马尔可夫模型轮廓比较工具(例如 HH-suite)搜索测试数据,该工具可以找到与训练数据[92] 远相关的序列。在常见的蛋白质结构被用作输入或输出的情况下,结构分类如 CATH [93] 或 ECOD [94] 可用于排除相似的折叠或同源蛋白质。类似的问题会影响预测蛋白质-配体结合亲和力的研究 [95]。
需要明确的是,数据泄漏不是任何特定类型数据的固有问题,而是在训练和评估机器学习模型时如何使用数据的问题。研究人员期望经过训练的模型能够在与训练集相似的数据上产生非常好的结果。当在某些基准集上看起来准确的模型,在实际上与训练集不同的新数据上表现不佳时,就可能发生了数据泄漏问题;换句话说,该模型没有泛化,可能是因为它没有对变量之间的真实关系进行建模,而是记住了数据中存在的隐藏关联。
由于审稿人的严格要求,一些学术期刊现在开始要求在考虑发表论文之前进行严格的基准测试。如果没有适当的测试,模型的性能很可能无法代表未见数据的真实性能,这会削弱用户对模型的信心。更糟糕的是,未来研究的作者可能会被误导,认为不充分的测试是有道理的,因为它已经出现在(可能是已经发表的)数篇经过同行评审的文章中。正如方框2中提到的,作者、同行评审和期刊编辑都有责任确保避免数据泄露。故意保留这些类型的错误,比起在实验结束时伪造数据,确实是半斤八两。
模型的可解释性。通常情况下,生物学家想知道为什么特定模型会做出特定预测(即模型响应输入数据的哪些特征以及如何响应)以及为什么它在某些情况下有效,而在其它情况下无效。换句话说,生物学家通常对发现负责建模输出的机制和因素感兴趣,而不仅仅对如前所述的准确建模。解释模型的能力取决于使用的机器学习方法和输入数据。对于非神经网络方法来说,解释通常更容易,因为这些方法具有更适合直接有意义解释的特征集,并且通常具有较少的可学习参数。例如,在简单线性回归模型的情况下,分配给每个输入特征的参数直接表明该特征如何影响预测。
训练非神经网络方法的低成本意味着进行消融研究(ablation study)是可取的,其中测量去除输入的定义特征对性能的影响。消融研究可以揭示哪些特征对手头的建模任务最有用,并且是一种可能发现更强大、更有效和可解释的模型的方法。
解释神经网络(尤其是深度神经网络)通常要困难得多,因为模型中经常有大量的输入特征和参数。识别输入图像中对特定分类最负责的区域仍然是有可能的,例如,通过构建显著图(saliency map) [28] 。尽管显著图显示了图像的哪些区域是重要的,但可能更难以确定这些位置的哪些数据属性是负责预测的,特别是当输入(例如图像和文本)不容易被人类解释时。尽管如此,显著图和类似概念可以用作“健全性检查”,以确保模型确实在查看图像的相关部分。这可以帮助避免模型进行意外连接的情况,例如,根据图像角落的医院或部门标签,而非图像本身的医学内容,对医学图像进行分类 [96]。生成对抗性示例(GAN)或合成输入可能导致神经网络产生自信的错误预测,但也是可以提供有关哪些特征最常用于预测的信息的好方法 [97]。例如,CNN 经常使用纹理(例如动物毛皮中的条纹)来对图像中的对象进行分类,而人类主要使用形状 [51]。
方框2 | 评估使用机器学习的文章
以下是在阅读或审阅使用机器学习处理生物数据的文章时需要考虑的一些问题。即使不一定得到有效回答,记住这些问题也是有用的,并且这些问题可以用作与机器学习背景合作者进行讨论的基础。有惊人数量的文章并不能达到这些标准[148]。
是否充分描述了数据集?
研究人员应提供构建数据集的完整步骤,最好使用在网页中永久保留的数据集或摘要数据。根据我们的经验,对机器学习方法进行全面描述,但对数据的描述却含糊其辞,是危险信号(red flag)。如果使用的数据集是标准数据集,或来自另一项研究,则应在文章中明确说明。
测试集是否确凿?
根据生物应用挑战部分中的讨论,检查测试集是否足以对所调查的属性进行基准测试。训练集和测试集之间不应该有数据泄漏,测试集应该足够大以提供可靠的结果,并且测试集应该覆盖该工具的用户可能的使用范围。研究人员同样应该详细讨论训练集和测试集的组成和大小。作者有责任确保已采取所有步骤以避免数据泄露,并且应在文章中描述这些步骤及其背后的基本原理。学术期刊编辑和审稿人还应该确保这些任务已经按照良好的标准执行,而非仅靠论文作者自觉。
模型选择是否合理?
研究人员应给出选择机器学习方法的理由。使用神经网络的理由是因为它们适用于当前数据和问题,而非仅仅是因为其他人都在使用。学术界应鼓励讨论已尝试但无效的模型,因为它可以帮助其他研究人员;很多时候,一个复杂的模型没有对最终得到该模型所需的不可避免的反复试验进行任何讨论,但依然得以发表。
该方法是否与其他方法进行了比较?
应该将一种新方法与表现出良好性能并被广泛使用的现有方法进行比较。理想情况下,应比较使用各种模型类型的方法,这样有助于解释结果。令人惊讶的是,许多复杂模型在性能上其实通过简单的回归方法就可以匹配。
结果好得令人难以置信吗?
超过 99% 准确率的声明,在生物学的机器学习文章中并不少见。通常,这是测试出现问题的迹象,而不是惊人的突破。作者和审稿人都应该注意这一点。
方法可用吗?
至少,想要使用文章中训练好模型的科研人员,应该能够基于网页服务或代码文件运行一次预测。理想情况下,至少在永久 URL 和通用许可下,应该提供源代码和经过训练的模型[149,150]。使训练代码可用,也是理想情况下该有的,因为这进一步提高了文章的可重复性并允许其他研究人员以该方法为基础,而无需从头开始。期刊应该在这里承担一些责任,以确保这成为常态。
保护隐私的机器学习。一些生物数据,尤其是人类基因组数据和商业敏感的药物数据,具有数据隐私方面的影响。在数据隐私的背景下,研究人员已经做出了许多努力,来实现数据共享和机器学习模型的分布式训练。例如,现代密码技术允许在数据和结果可证明是安全的情况下,训练药物-靶标相互作用模型[98]。与临床试验中的真实参与者非常相似的模拟合成参与者,可以在不泄露识别数据的前提下,得出对真实参与者准确的结果[99]。研究人员已经开发了一些算法,可以使用存储在不同位置的数据,进行有效的联合模型训练 [100]。
跨学科合作的必要性。除非使用公开可用的数据,否则一个研究组很少会同时拥有专业知识和资源——既可以为机器学习研究收集数据,又可以有效地应用最合适的机器学习方法。实验生物学家与计算机科学家合作十分常见,而且通常会取得很好的结果。然而,在这种合作中,重要的是每一方都了解对方的一些工作知识。特别是计算机科学家应该努力了解数据——例如预期的噪声程度和可重复性——同时生物学家应该了解所使用的机器学习算法的局限性。建立这样的理解需要时间和精力,但对于防止不良模型和误导性结果的无意传播很重要。
在可预见的未来,机器学习在生物学研究中的使用越来越多的趋势,看起来也将继续下去。方法论、软件和硬件的重要进步使这种吸收增加成为可能,所有方向都在不断发展。许多大型科技公司正在利用他们的技术专长和大量资源来协助学术研究人员,甚至通过创新的机器学习策略进行生物学研究。然而,迄今为止,大多数成功来自将其他领域开发的算法直接应用于生物数据。例如,CNN 和 RNN 分别更多应用于图像分析(用于人脸识别或自动驾驶汽车)和自然语言处理等应用。生物学机器学习最令人兴奋的前景之一,是专门针对生物学数据和生物学问题量身定制的算法 [101,102] 。如果可以利用生物系统的已知结构并使用神经网络来学习未知部分,就可以用更易于解释且对新数据更稳健的更简单的模型,来取代参数化越来越严重的模型 [103]。应用包括生物反应系统和药代动力学,在这些应用中可以使用已知微分方程系统。这也将有助于从预测机器学习转向可以创建新实体的生成模型,例如设计具有新结构和功能的蛋白质 [104,105]。
随着有用架构和输入数据类型种类的增加,可微编程的范式正在从深度学习领域出现 [106]。可微编程是使用自动微分(训练神经网络的核心概念)来计算梯度和改进任何所需算法中的参数。这显示了蛋白质结构预测中生物系统物理模型的前景 [63,107],以及学习分子动力学模拟的力场参数 [108,109]。可微软件包(如 JAX [110])和针对特定生物学领域的软件包(如 Selene [111]、Janggu [112] 和 JAX MD [113])的开发将有助于此类方法的开发。
使用机器学习进行生物数据分析的进展也得益于在公开可用的存储库中存放训练好的模型。这样,研究类似问题的研究人员可以使用这些模型而无需训练,并且可以使用各种不同的模型,只需极少的工作即可在它们之间切换[114]。该领域还出现了一种扩展——自动化机器学习管道,无需用户输入即可训练和调整各种模型并返回性能最佳的模型。这些管道可以帮助非专家训练模型[115]。然而,这些资源不能替代对所用方法的透彻理解,对于选择适当的归纳偏差和解释模型的预测很重要。至于自动化机器学习是否已经可靠和灵活到允许实验人员独立地常规使用复杂的机器学习算法,或者说,机器学习专业知识是否仍然是必需的,还有待未来观察。
如前所述,模型的严格验证和不同模型的比较具有挑战性,但对于确定性能最佳的模型和为未来的研究方向提供信息仍然是必要的。为了使该领域取得进展,有必要开发基准数据集和验证任务 [116],例如 Protein Net [117]、ATOM3D [118] 和 TAPE [119],并使其得到广泛应用。当然,可能会发生针对特定基准的过度优化,所以重要的是需要研究人员抵制这种能使他们的结果看起来更好的诱惑。诸如 CASP [120] 和功能注释的批判性评估 [121] 等盲评估,将继续在评估哪些模型表现最佳方面,发挥重要作用。
总体而言,生物数据的多样性使得机器学习很难为其生物学中的应用提供通用指南。因此,本文的目标是为生物学家概述可用的不同方法,并为他们提供一些关于如何在他们的数据上进行有效机器学习的预判。当然,机器学习并不适合所有问题,知道它什么时候变为劣势同样重要:在没有足够的数据时、在需要理解而非预测时,或者在不清楚如何评估性能时。机器学习在生物学中何时能发挥最大效能的界限,仍有待探索,并将根据可用实验数据的性质和数量继续变化。但不可否认的是,机器学习已经对生物学产生了巨大影响,并将继续如此。
1. Ching, T. et al. Opportunities and obstacles for deep learning in biology and medicine. J. R. Soc. Interface 15, 20170387 (2018).
2.Mitchell, T. M. Machine Learning (McGraw Hill, 1997).
3.Goodfellow, I., Bengio Y. & Courville, A. Deep Learning (MIT Press, 2016).
4. Libbrecht, M. W. & Noble, W. S. Machine learning applications in genetics and genomics. Nat. Rev. Genet.16, 321–332 (2015).
5. Zou, J. et al. A primer on deep learning in genomics.Nat. Genet. 51, 12–18 (2019).
6.Myszczynska, M. A. et al. Applications of machine learning to diagnosis and treatment of neurodegenerative diseases. Nat. Rev. Neurol. 16,440–456 (2020).
7.Yang, K. K., Wu, Z. & Arnold, F. H. Machine- learning-guided directed evolution for protein engineering. Nat. Methods 16, 687–694 (2019).
8.Tarca, A. L., Carey, V. J., Chen, X.-W., Romero, R.& Drăghici, S. Machine learning and its applications to biology. PLoS Comput. Biol. 3, e116 (2007).This is an introduction to machine learning concepts and applications in biology with a focus on traditional machine learning methods.
9.Silva, J. C. F., Teixeira, R. M., Silva, F. F.,Brommonschenkel, S. H. & Fontes, E. P. B. Machine learning approaches and their current application in plant molecular biology: a systematic review.Plant. Sci. 284, 37–47 (2019).
10. Kandoi, G., Acencio, M. L. & Lemke, N. Prediction of druggable proteins using machine learning and systems biology: a mini- review. Front. Physiol. 6, 366 (2015).
11. Marblestone, A. H., Wayne, G. & Kording, K. P. Toward an integration of deep learning and neuroscience.Front. Comput. Neurosci. 10, 94 (2016).
12. Jiménez- Luna, J., Grisoni, F. & Schneider, G.Drug discovery with explainable artificial intelligence. Nat. Mach. Intell. 2, 573–584 (2020).
13. Buchan, D. W. A. & Jones, D. T. The PSIPRED Protein Analysis Workbench: 20 years on. Nucleic Acids Res.47, W402–W407 (2019).
14. Kelley, D. R., Snoek, J. & Rinn, J. L. Basset: learning the regulatory code of the accessible genome with deep convolutional neural networks. Genome Res. 26,990–999 (2016).
15. Altman, N. & Krzywinski, M. Clustering. Nat. Methods 14, 545–546 (2017).
16. Hopf, T. A. et al. Mutation effects predicted from sequence co- variation. Nat. Biotechnol. 35, 128–135(2017).
17. Zhang, Z. et al. Predicting folding free energy changes upon single point mutations. Bioinformatics 28,664–671 (2012).
18. Pedregosa, F. et al. Scikit- learn: machine learning in python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
19. Kuhn, M. Building predictive models in r using the caret package. J. Stat. Softw. 28, 1–26 (2008).
20. Blaom, A. D. et al. MLJ: a Julia package for composable machine learning. J. Open Source Softw.5, 2704 (2020).
21. Jones, D. T. Setting the standards for machine learning in biology. Nat. Rev. Mol. Cell Biol. 20, 659–660(2019).
22. Alipanahi, B., Delong, A., Weirauch, M. T. & Frey, B. J. Predicting the sequence specificities of DNA- and RNA- binding proteins by deep learning.Nat. Biotechnol. 33, 831–838 (2015).
23. Senior, A. W. et al. Improved protein structure prediction using potentials from deep learning.Nature 577, 706–710 (2020).
Technology company DeepMind entered the CASP13 assessment in protein structure prediction and its method using deep learning was the most accurate of the methods entered.
24. Esteva, A. et al. Dermatologist- level classification of skin cancer with deep neural networks. Nature 542,115–118 (2017).
25. Tegunov, D. & Cramer, P. Real- time cryo- electron microscopy data preprocessing with Warp.Nat. Methods 16, 1146–1152 (2019).
26. LeCun, Y., Bengio, Y. & Hinton, G. Deep learning. Nature 521, 436–444 (2015). This is a review of deep learning by some of the major figures in the deep learning revolution.
27. Hastie T., Tibshirani R., Friedman J. The elements of statistical learning: data mining, inference, and prediction. 2nd Edn. (Springer Science & Business Media; 2009).
28. Adebayo, J. et al. Sanity checks for saliency maps. NeurIPS https://arxiv.org/abs/1810.03292 (2018).
29. Gal, Y. & Ghahramani, Z. Dropout as a Bayesian approximation: representing model uncertainty in deep learning. ICML 48, 1050–1059 (2016).
30. Smith, A. M. et al. Standard machine learning approaches outperform deep representation learning on phenotype prediction from transcriptomics data. BMC Bioinformatics 21, 119 (2020).
31. Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B. 58, 267–288(1996).
32. Zou, H. & Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B. 67,301–320 (2005).
33. Noble, W. S. What is a support vector machine? Nat. Biotechnol. 24, 1565–1567 (2006).
34. Ben- Hur, A. & Weston, J. A user’s guide to support vector machines. Methods Mol. Biol. 609, 223–239(2010).
35. Ben- Hur, A., Ong, C. S., Sonnenburg, S., Schölkopf, B.& Rätsch, G. Support vector machines and kernels for computational biology. PLoS Comput. Biol. 4,e1000173 (2008). This is an introduction to SVMs with a focus on biological data and prediction tasks.
36. Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants.Nat. Genet. 46, 310–315 (2014).
37. Driscoll, M. K. et al. Robust and automated detection of subcellular morphological motifs in 3D microscopy images. Nat. Methods 16, 1037–1044 (2019).
38. Bzdok, D., Krzywinski, M. & Altman, N. Machine learning: supervised methods. Nat. Methods 15, 5–6 (2018).
39. Wang, C. & Zhang, Y. Improving scoring- docking-screening powers of protein- ligand scoring functions using random forest. J. Comput. Chem. 38, 169–177(2017).
40. Zeng, W., Wu, M. & Jiang, R. Prediction of enhancer- promoter interactions via natural language processing. BMC Genomics 19, 84 (2018).
41. Olson, R. S., Cava, W. L., Mustahsan, Z., Varik, A. & Moore, J. H. Data- driven advice for applying machine learning to bioinformatics problems. Pac. Symp. Biocomput. 23, 192–203 (2018).
42. Rappoport, N. & Shamir, R. Multi- omic and multi- view clustering algorithms: review and cancer benchmark.Nucleic Acids Res. 47, 1044 (2019).
43. Steinegger, M. & Söding, J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat. Biotechnol. 35,1026–1028 (2017).
44. Jain, A. K. Data clustering: 50 years beyond K- means.Pattern Recognit. Lett. 31, 651–666 (2010).
45. Ester M., Kriegel H.-P., Sander J., Xu X. A density- based algorithm for discovering clusters in large spatial databases with noise. KDD‘96 Proc. Second Int. Conf. Knowl. Discov. Data Mining. 96, 226–231 (1996).
46. Nguyen, L. H. & Holmes, S. Ten quick tips for effective dimensionality reduction. PLoS Comput. Biol. 15,e1006907 (2019).
47. Moon, K. R. et al. Visualizing structure and transitions in high- dimensional biological data. Nat. Biotechnol.37, 1482–1492 (2019).
48. van der Maaten, L. & Hinton, G. Visualizing data using t- SNE. J. Mach. Learn. Res. 9, 2579–2605 (2008).
49. Kobak, D. & Berens, P. The art of using t- SNE for single- cell transcriptomics. Nat. Commun. 10, 5416 (2019).
This article provides a discussion and tips for using t- SNE as a dimensionality reduction technique on single- cell transcriptomics data.
50. Crick, F. The recent excitement about neural networks.Nature 337, 129–132 (1989).
51. Geirhos, R. et al. Shortcut learning in deep neural networks. Nat. Mach. Intell. 2, 665–673 (2020). This article discusses a common problem in deep learning called ‘shortcut learning’, where the model uses decision rules that do not transfer to real- world data.
52. Qian, N. & Sejnowski, T. J. Predicting the secondary structure of globular proteins using neural network models. J. Mol. Biol. 202, 865–884 (1988).
53. deFigueiredo, R. J. et al. Neural- network-based classification of cognitively normal, demented, Alzheimer disease and vascular dementia from single photon emission with computed tomography image data from brain. Proc. Natl Acad. Sci. USA 92, 5530–5534 (1995).
54. Mayr, A., Klambauer, G., Unterthiner, T. & Hochreiter, S.DeepTox: toxicity prediction using deep learning.Front. Environ. Sci. 3, 80 (2016).
55. Yang, J. et al. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl Acad. Sci. USA 117, 1496–1503 (2020).
56. Xu, J., Mcpartlon, M. & Li, J. Improved protein structure prediction by deep learning irrespective of co-evolution information. Nat. Mach. Intell. 3,601–609 (2021).
57. Poplin, R. et al. A universal SNP and small- indel variant caller using deep neural networks.Nat. Biotechnol. 36, 983–987 (2018).
58. Fudenberg, G., Kelley, D. R. & Pollard, K. S. Predicting 3D genome folding from DNA sequence with Akita.Nat. Methods 17, 1111–1117 (2020).
59. Zeng, H., Edwards, M. D., Liu, G. & Gifford, D. K.Convolutional neural network architectures for predicting DNA- protein binding. Bioinformatics 32,i121–i127 (2016).
60. Yao, R., Qian, J. & Huang, Q. Deep- learning with synthetic data enables automated picking of cryo- EM particle images of biological macromolecules.Bioinformatics 36, 1252–1259 (2020).
61. Si, D. et al. Deep learning to predict protein backbone structure from high- resolution cryo- EM density maps.Sci. Rep. 10, 4282 (2020).
62. Poplin, R. et al. Prediction of cardiovascular risk factors from retinal fundus photographs via deep learning. Nat. Biomed. Eng. 2, 158–164 (2018).
63. AlQuraishi, M. End- to-end differentiable learning of protein structure. Cell Syst. 8, 292–301.e3 (2019).
64. Heffernan, R., Yang, Y., Paliwal, K. & Zhou, Y.Capturing non- local interactions by long short- term memory bidirectional recurrent neural networks for improving prediction of protein secondary structure,backbone angles, contact numbers and solvent accessibility. Bioinformatics 33, 2842–2849 (2017).
65. Müller, A. T., Hiss, J. A. & Schneider, G. Recurrent neural network model for constructive peptide design.J. Chem. Inf. Model. 58, 472–479 (2018).
66. Choi, E., Bahadori, M. T., Schuetz, A., Stewart, W. F.& Sun, J. Doctor AI: predicting clinical events via recurrent neural networks. JMLR Workshop Conf.Proc. 56, 301–318 (2016).
67. Quang, D. & Xie, X. DanQ: a hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res. 44,e107 (2016).
68. Alley, E. C., Khimulya, G., Biswas, S., AlQuraishi, M.& Church, G. M. Unified rational protein engineering with sequence- based deep representation learning.Nat. Methods 16, 1315–1322 (2019).
69. Vaswani, A. et al. Attention is all you need.arXiv https://arxiv.org/abs/1706.03762 (2017).
70. Elnaggar, A. et al. ProtTrans: towards cracking the language of life’s code through self-supervised deep learning and high performance computing. arXiv https://arxiv.org/abs/2007.06225 (2020).
71. Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021).
72. Battaglia, P. W. et al. Relational inductive biases, deep learning, and graph networks. arXiv https://arxiv.org/ abs/1806.01261 (2018).
73. Stokes, J. M. et al. A deep learning approach to antibiotic discovery. Cell 181, 475–483 (2020). In this work, a deep learning model predicts antibiotic activity, with one candidate showing broad- spectrum antibiotic activities in mice.
74. Gainza, P. et al. Deciphering interaction fingerprints from protein molecular surfaces using geometric deep learning. Nat. Methods 17, 184–192 (2020).
75. Strokach, A., Becerra, D., Corbi- Verge, C., Perez- Riba, A.& Kim, P. M. Fast and flexible protein design using deep graph neural networks. Cell Syst. 11, 402–411.e4(2020).
76. Gligorijevic, V. et al. Structure-based function prediction using graph convolutional networks. Nat. Commun. 12, 3168 (2021).
77. Zitnik, M., Agrawal, M. & Leskovec, J. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics 34, i457–i466 (2018).
78. Veselkov, K. et al. HyperFoods: machine intelligent mapping of cancer- beating molecules in foods.Sci. Rep. 9, 9237 (2019).
79. Fey, M. & Lenssen, J. E. Fast graph representation learning with PyTorch geometric. arXiv https://arxiv.org/abs/1903.02428 (2019).
80. Zhavoronkov, A. et al. Deep learning enables rapid identification of potent DDR1 kinase inhibitors. Nat. Biotechnol. 37, 1038–1040 (2019).
81. Wang, Y. et al. Predicting DNA methylation state of CpG dinucleotide using genome topological features and deep networks. Sci. Rep. 6, 19598(2016).
82. Linder, J., Bogard, N., Rosenberg, A. B. & Seelig, G.A generative neural network for maximizing fitness and diversity of synthetic DNA and protein sequences.Cell Syst. 11, 49–62.e16 (2020).
83. Greener, J. G., Moffat, L. & Jones, D. T. Design of metalloproteins and novel protein folds using variational autoencoders. Sci. Rep. 8, 16189(2018).
84. Wang, J. et al. scGNN is a novel graph neural network framework for single- cell RNA- Seq analyses. Nat. Commun. 12, 1882 (2021).
85. Paszke, A. et al. PyTorch: an imperative style, high- performance deep learning library. Adv. Neural Inf. Process. Syst. 32, 8024–8035 (2019).
86. Abadi M. et al. Tensorflow: a system for large- scale machine learning. 12th USENIX Symposium on Operating Systems Design and Implementation.265–283 (USENIX, 2016).
87. Wei, Q. & Dunbrack, R. L. Jr The role of balanced training and testing data sets for binary classifiers in bioinformatics. PLoS ONE 8, e67863 (2013).
88. Walsh, I., Pollastri, G. & Tosatto, S. C. E. Correct machine learning on protein sequences: a peer-reviewing perspective. Brief. Bioinform 17, 831–840(2016).
This article discusses how peer reviewers can assess machine learning methods in biology, and by extension how scientists can design and conduct such studies properly.
89. Schreiber, J., Singh, R., Bilmes, J. & Noble, W. S.A pitfall for machine learning methods aiming to predict across cell types. Genome Biol. 21, 282(2020).
90. Chothia, C. & Lesk, A. M. The relation between the divergence of sequence and structure in proteins. EMBO J. 5, 823–826 (1986).
91. Söding, J. & Remmert, M. Protein sequence comparison and fold recognition: progress and good- practice benchmarking. Curr. Opin. Struct. Biol.21, 404–411 (2011).
92. Steinegger, M. et al. HH- suite3 for fast remote homology detection and deep protein annotation. BMC Bioinformatics 20, 473 (2019).
93. Sillitoe, I. et al. CATH: expanding the horizons of structure- based functional annotations for genome sequences. Nucleic Acids Res. 47, D280–D284(2019).
94. Cheng, H. et al. ECOD: an evolutionary classification of protein domains. PLoS Comput. Biol. 10, e1003926(2014).
95. Li, Y. & Yang, J. Structural and sequence similarity makes a significant impact on machine- learning-based scoring functions for protein- ligand interactions.J. Chem. Inf. Model. 57, 1007–1012 (2017).
96. Zech, J. R. et al. Variable generalization performance of a deep learning model to detect pneumonia in chest radiographs: a cross- sectional study. PLoS Med. 15,e1002683 (2018).
97. Szegedy, C. et al. Intriguing properties of neural networks. arXiv https://arxiv.org/abs/1312.6199 (2014).
98. Hie, B., Cho, H. & Berger, B. Realizing private and practical pharmacological collaboration. Science 362,347–350 (2018).
99. Beaulieu- Jones, B. K. et al. Privacy- preserving generative deep neural networks support clinical data sharing. Circ. Cardiovasc. Qual. Outcomes 12,e005122 (2019).
100. Konečný, J., Brendan McMahan, H., Ramage, D.& Richtárik, P. Federated optimization: distributed machine learning for on-device intelligence. arXiv https://arxiv.org/abs/1610.02527 (2016).
101. Pérez, A., Martínez- Rosell, G. & De Fabritiis, G.Simulations meet machine learning in structural biology. Curr. Opin. Struct. Biol. 49, 139–144 (2018).
102. Noé, F., Olsson, S., Köhler, J. & Wu, H. Boltzmann generators: sampling equilibrium states of many-body systems with deep learning. Science 365, 6457(2019).
103. Shrikumar, A., Greenside, P. & Kundaje, A. Reverse-complement parameter sharing improves deep learning models for genomics. bioRxiv https://www.biorxiv.org/content/10.1101/103663v1 (2017).
104. Lopez, R., Gayoso, A. & Yosef, N. Enhancing scientific discoveries in molecular biology with deep generative models. Mol. Syst. Biol. 16, e9198 (2020).
105. Anishchenko, I., Chidyausiku, T. M., Ovchinnikov, S.,Pellock, S. J. & Baker, D. De novo protein design by deep network hallucination. bioRxiv https://doi.org/10.1101/2020.07.22.211482 (2020).
106. Innes, M. et al. A differentiable programming system to bridge machine learning and scientific computing. arXiv https://arxiv.org/abs/1907.07587 (2019).
107. Ingraham J., Riesselman A. J., Sander C., Marks D. S.Learning protein structure with a differentiable simulator.ICLR https://openreview.net/forum?id=Byg3y3C9Km(2019).
108. Jumper, J. M., Faruk, N. F., Freed, K. F. & Sosnick, T. R.Trajectory- based training enables protein simulations with accurate folding and Boltzmann ensembles in cpu- hours. PLoS Comput. Biol. 14, e1006578 (2018).
109. Wang, Y., Fass, J. & Chodera, J. D. End-to-end differentiable molecular mechanics force field construction. arXiv http://arxiv.org/abs/2010.01196(2020).
110. Bradbury, J. et al. JAX: composable transformations of Python+NumPy programs. GitHub http://github.com/google/jax (2018).
111. Chen, K. M., Cofer, E. M., Zhou, J. & Troyanskaya, O. G.Selene: a PyTorch- based deep learning library for sequence data. Nat. Methods 16, 315–318 (2019).
This work provides a software library based on PyTorch providing functionality for biological sequences.
112. Kopp, W., Monti, R., Tamburrini, A., Ohler, U.& Akalin, A. Deep learning for genomics using Janggu. Nat. Commun. 11, 3488 (2020).
113. Schoenholz, S. S. & Cubuk, E. D. JAX, M.D.: end-to-end differentiable, hardware accelerated, molecular dynamics in pure Python. arXiv https://arxiv.org/abs/1912.04232 (2019).
114. Avsec, Ž. et al. The Kipoi repository accelerates community exchange and reuse of predictive models for genomics. Nat. Biotechnol. 37, 592–600 (2019).
115. Isensee, F., Jaeger, P. F., Kohl, S. A. A., Petersen, J.& Maier- Hein, K. H. nnU- Net: a self- configuring method for deep learning- based biomedical image segmentation. Nat Methods 18, 203–211 (2020).
116. Livesey, B. J. & Marsh, J. A. Using deep mutational scanning to benchmark variant effect predictors and identify disease mutations. Mol. Syst. Biol. 16, e9380 (2020).
117. AlQuraishi, M. ProteinNet: a standardized data set for machine learning of protein structure.BMC Bioinformatics 20, 311 (2019).
118. Townshend, R. J. L. et al. ATOM3D: tasks on molecules in three dimensions. arXiv https://arxiv.org/abs/2012.04035 (2020).
119. Rao, R. et al. Evaluating protein transfer learning with TAPE. Adv. Neural. Inf. Process. Syst. 32, 9689–9701(2019).
120. Kryshtafovych, A., Schwede, T., Topf, M., Fidelis, K.& Moult, J. Critical assessment of methods of protein structure prediction (CASP) — round XIII. Proteins 87,1011–1020 (2019).
121. Zhou, N. et al. The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens. Genome Biol. 20, 244 (2019).
122. Munro, D. & Singh, M. DeMaSk: a deep mutational scanning substitution matrix and its use for variant impact prediction. Bioinformatics 36, 5322–5329(2020).
123. Haario, H. & Taavitsainen, V.-M. Combining soft and hard modelling in chemical kinetic models. Chemom.Intell. Lab. Syst. 44, 77–98 (1998).
124. Cozzetto, D., Minneci, F., Currant, H. & Jones, D. T.FFPred 3: feature- based function prediction for all gene ontology domains. Sci. Rep. 6, 31865 (2016).
125. Nugent, T. & Jones, D. T. Transmembrane protein topology prediction using support vector machines.BMC Bioinformatics 10, 159 (2009).
126. Bao, L., Zhou, M. & Cui, Y. nsSNPAnalyzer: identifying disease- associated nonsynonymous single nucleotide polymorphisms. Nucleic Acids Res. 33, W480–W482(2005).
127. Li, W., Yin, Y., Quan, X. & Zhang, H. Gene expression value prediction based on XGBoost algorithm. Front.Genet. 10, 1077 (2019).
128. Zhang, Y. & Skolnick, J. SPICKER: a clustering approach to identify near- native protein folds. J. Comput. Chem.30, 865–871 (2004).
129. Teodoro, M. L., Phillips, G. N. Jr & Kavraki, L. E.Understanding protein flexibility through dimensionality reduction. J. Comput. Biol. 10,617–634 (2003).
130. Schlichtkrull, M. et al. Modeling relational data with graph convolutional networks. arXiv https://arxiv.org/abs/1703.06103 (2019).
131. Pandarinath, C. et al. Inferring single- trial neural population dynamics using sequential auto- encoders.Nat. Methods 15, 805–815 (2018).
132. Antczak, M., Michaelis, M. & Wass, M. N.Environmental conditions shape the nature of a minimal bacterial genome. Nat. Commun. 10, 3100 (2019).
133. Sun, T., Zhou, B., Lai, L. & Pei, J. Sequence- based prediction of protein protein interaction using a deep- learning algorithm. BMC Bioinformatics 18,277 (2017).
134. Hiranuma, N. et al. Improved protein structure refinement guided by deep learning based accuracy estimation. Nat. Commun. 12, 1340 (2021).
135. Pagès, G., Charmettant, B. & Grudinin, S. Protein model quality assessment using 3D oriented convolutional neural networks. Bioinformatics 35,3313–3319 (2019).
136. Pires, D. E. V., Ascher, D. B. & Blundell, T. L. DUET: a server for predicting effects of mutations on protein stability using an integrated computational approach.Nucleic Acids Res. 42, W314–W319 (2014).
137. Yuan, Y. & Bar- Joseph, Z. Deep learning for inferring gene relationships from single- cell expression data.Proc. Natl Acad. Sci. USA 116, 27151–27158 (2019).
138. Chen, L., Cai, C., Chen, V. & Lu, X. Learning a hierarchical representation of the yeast transcriptomic machinery using an autoencoder model. BMC Bioinformatics 17, S9 (2016).
139. Kantz, E. D., Tiwari, S., Watrous, J. D., Cheng, S.& Jain, M. Deep neural networks for classification of LC- MS spectral peaks. Anal. Chem. 91, 12407–12413(2019).
140. Dührkop, K. et al. SIRIUS 4: a rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 16, 299–302 (2019).
141. Liebal, U. W., Phan, A. N. T., Sudhakar, M., Raman, K. & Blank, L. M. Machine learning applications for mass spectrometry- based metabolomics. Metabolites 10,243 (2020).
142. Zhong, E. D., Bepler, T., Berger, B. & Davis, J. H. CryoDRGN: reconstruction of heterogeneous cryo- EM structures using neural networks. Nat. Methods 18,176–185 (2021).
143. Schmauch, B. et al. A deep learning model to predict RNA- Seq expression of tumours from whole slide images. Nat. Commun. 11, 3877 (2020).
144. Das, P. et al. Accelerated antimicrobial discovery via deep generative models and molecular dynamics simulations. Nat. Biomed. Eng. 5, 613–623 (2021).
145. Gligorijevic, V., Barot, M. & Bonneau, R. deepNF:deep network fusion for protein function prediction.Bioinformatics 34, 3873–3881 (2018).
146. Karpathy A. A recipe for training neural networks. https://karpathy.github.io/2019/04/25/recipe (2019).
147. Bengio, Y. Practical recommendations for gradient-based training of deep architectures. Lecture Notes Comput. Sci. 7700, 437–478 (2012).
148. Roberts, M. et al. Common pitfalls and recommendations for using machine learning to detect and prognosticate for COVID-19 using chest radiographs and CT scans. Nat. Mach. Intell. 3,199–217 (2021).
This study assesses 62 machine learning studies that analyse medical images for COVID-19 and none is found to be of clinical use, indicating the difficulties of training a useful model.
149. List, M., Ebert, P. & Albrecht, F. Ten simple rules for developing usable software in computational biology.PLoS Comput. Biol. 13, e1005265 (2017).
150. Sonnenburg, S. Ã., Braun, M. L., Ong, C. S. & Bengio, S.The need for open source software in machine learning.J. Mach. Learn. Res. 8, 2443–2466 (2007).
集智斑图顶刊论文速递栏目上线以来,持续收录来自Nature、Science等顶刊的最新论文,追踪复杂系统、网络科学、计算社会科学等领域的前沿进展。现在正式推出订阅功能,每周通过微信服务号「集智斑图」推送论文信息。扫描下方二维码即可一键订阅:

点击“阅读原文”,追踪复杂科学顶刊论文