随着单细胞转录组及空间转录组技术的成熟,研究者需要将多个来源、不同条件下的数据进行整合,便于具有生物学意义数据的挖掘。近日,来自浙江树人大学树兰国际医学院的王力飞联合北京师范大学系统科学学院张江组,以及中国科学院北京基因组研究所蔡军组,提出了基于变分自编码器(VAE)的生成式模型 inClust,能够在有监督、半监督及无监督模式下整合分析转录组数据,其表现优于现有方法,且可灵活地进行数据集成、分解、查询等多项任务。
研究领域:生命复杂系统,单细胞转录组,机器学习,生成模型,变分自编码器
论文题目:
A deep generative framework with embedded vector arithmetic and classifier for sample generation, label transfer, and clustering of single-cell data
https://www.cell.com/cell-reports-methods/pdf/S2667-2375(23)00202-3.pdf
人体复杂性的根源在于微观层面上交互的不同类型的细胞。这些细胞有着相同的 DNA,但不同类型细胞有着不同的基因表达谱(即不同基因分别有多少份拷贝被翻译成具有蛋白质,从而执行对应的功能)。正是由于不同的细胞有着不同的基因表达谱,生物体才会在同一份蓝图的指引下谱写出千姿百态的乐章。
转录组测序可检测样本中各种基因的表达量。传统的检测技术分辨率不够高,不能达到单个细胞的精度,其应用受限。而单细胞转录组可检测到单个细胞中的基因表达量,并通过在不同类型(如是否患病)样本间的比较,找出具有生物学意义的差异、共表达及调控模式。近年来新出现的空间转录组技术,能够记录样本来源的空间坐标,即每一个转录组数据带有其所属位置的二维坐标信息,提供了更加丰富的信息。
然而,转录组数据是一种复杂的数据,其测量值会受到生物以及非生物因素的影响。比如不同批次、不同实验条件以及不同测序技术的数据,会由于各种差异导致不同来源的数据不能整合到一起使用。如图1的聚类结果所示,其中每个点代表一个细胞,图1b中的颜色代表细胞类型,其中不同颜色对应不同细胞类型,然而相同类型的细胞并没有被聚在一起,反而在图1c中,不同批次的数据聚类后明显地聚成一簇。
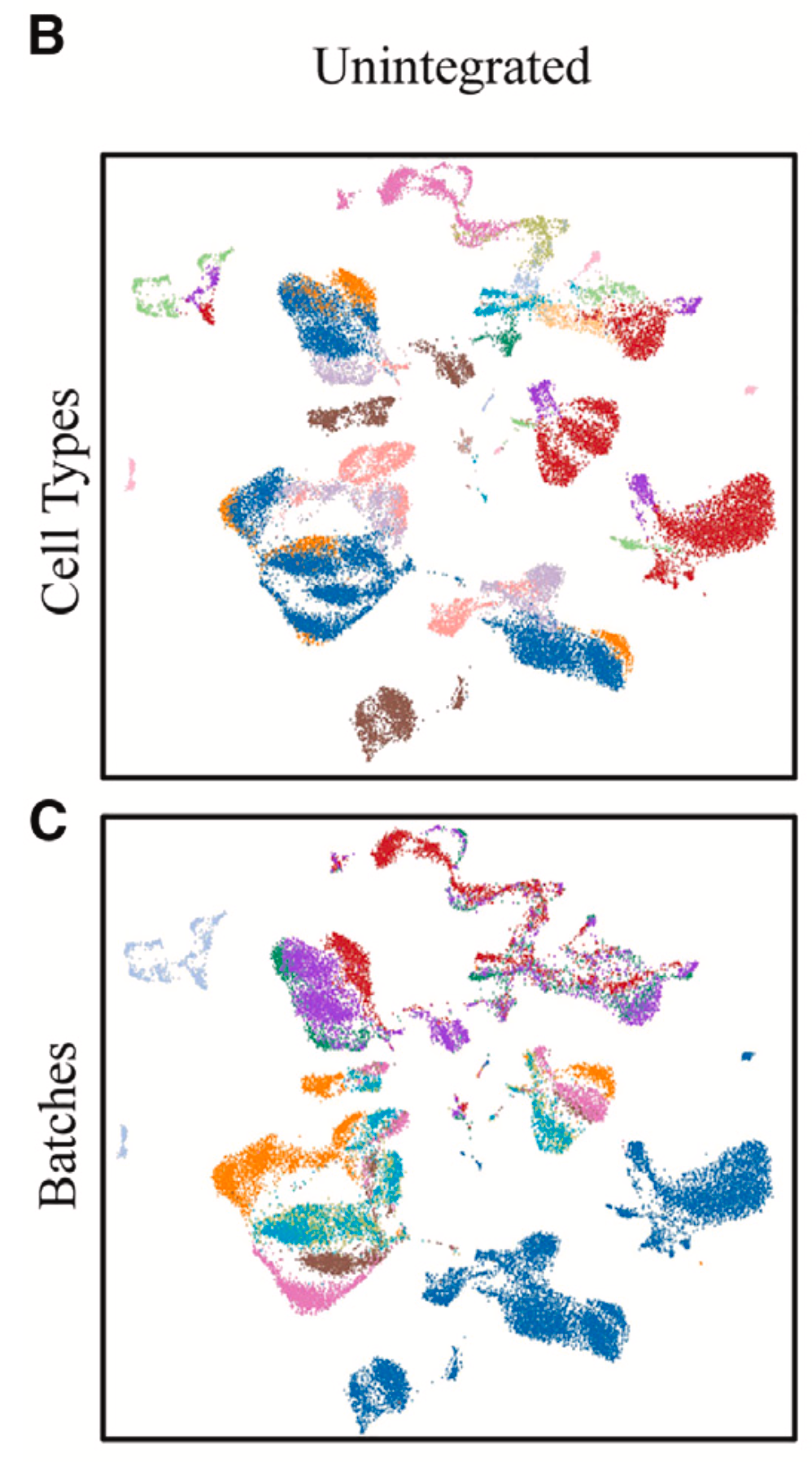
图1:未进行数据整合前的多批次单细胞转录组聚类情况
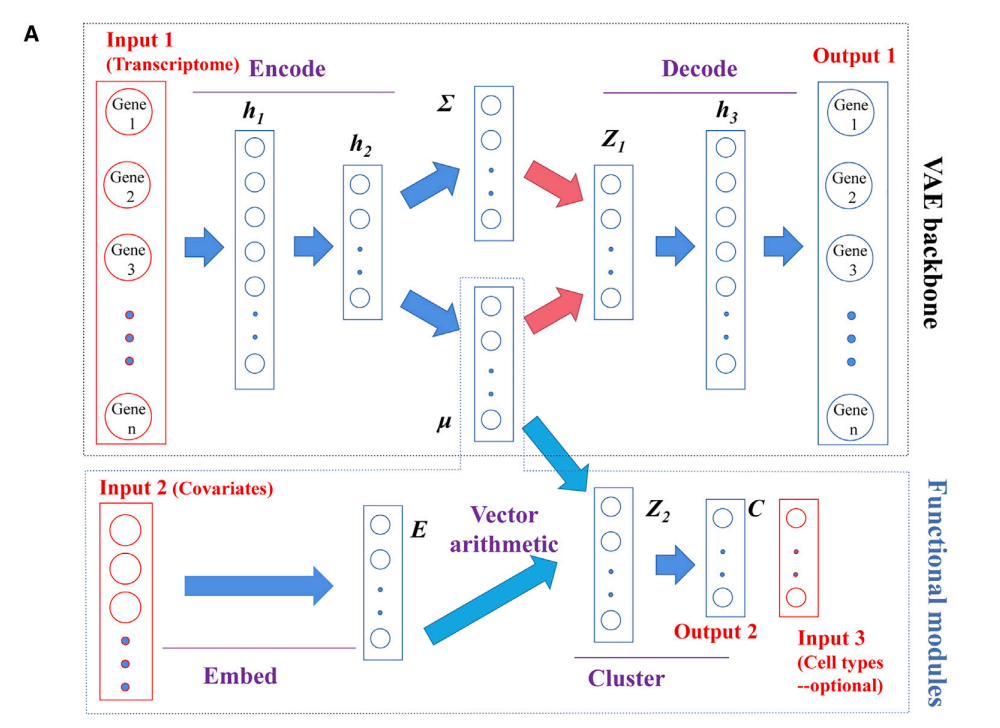
为了解决上述数据整合问题,已有多种成熟的方法,然而这些方法大多是为了解决单一问题提出的。inClust 作为一种灵活鲁棒的生成模型,可以执行对样本的辅助信息进行嵌入,在隐空间中进行数值运算、聚类、查询及批次整合等多种任务。另外,在数据整合后,inClust还能解耦数据,将复杂的转录组数据分解为简单数据的组合,完成数据的有意义的拆分,并能仿真生成新场景下的数据。
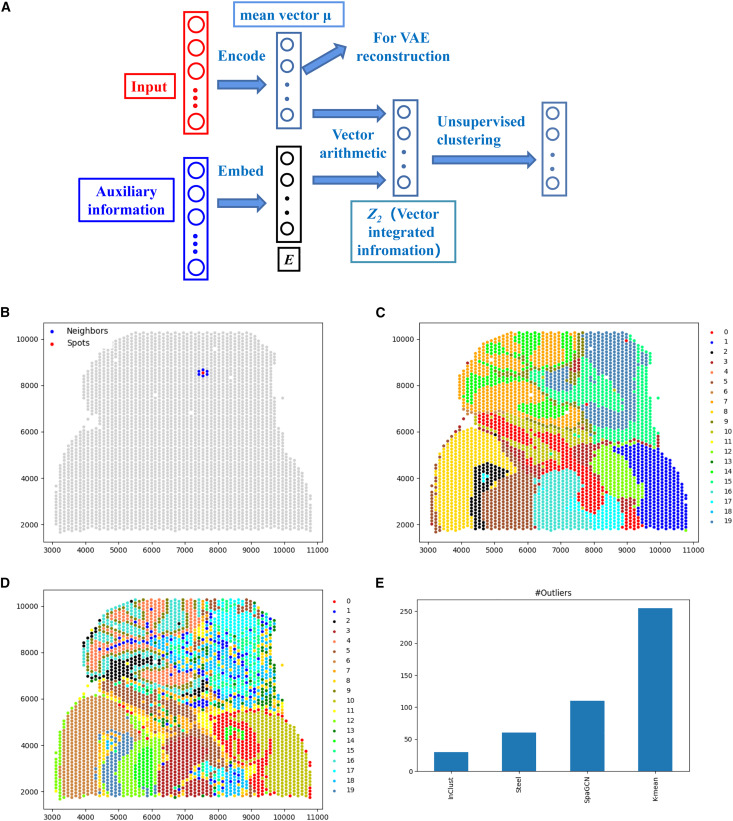
inClust 基于变分自编码器(VAE),其输入分为样本表达向量,样本的辅助信息(如样本的批次信息),以及每个样本对应的细胞类型标签(该输入不是必须)。运行时,模型同时将表达向量和辅助信息分别编码(嵌入)到低维空间,并在低维空间中使用向量运算整合信息,最后分类器将整合信息后的向量进行分类。如果输入中有细胞类型标签时,该输入可作为约束,监督分类器输出的结果。
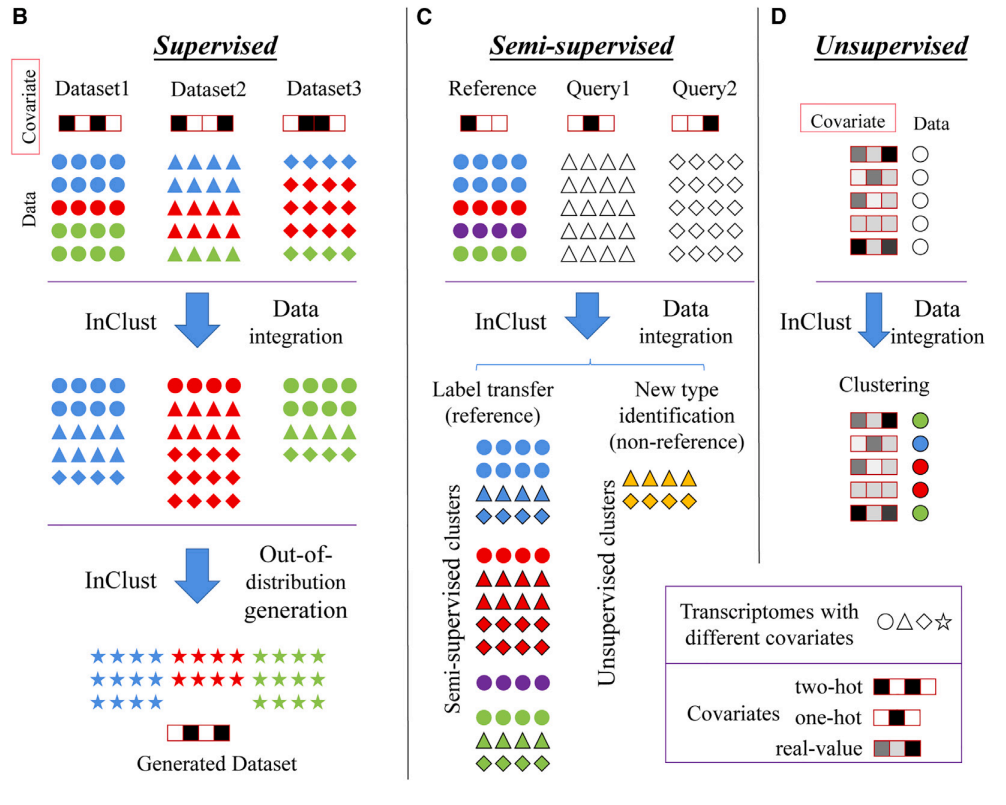
当所有数据都有细胞类型标签时,inClust 用来生成与原数据分布不同的仿真数据(图3b);而当只有部分数据包含细胞类型标签时,inClust 可以半监督的形式运行(图3c),根据有标注数据(参考集)进行标签迁移,预测未标注数据(查询集)的类型;而在所有数据都没有标签时,inClust的无监督模式可对数据进行整合及聚类。
在数据整合前,相同细胞类型的细胞因为批次效应,不能聚集到一起(图4B,C第一列)。经过inClust整合后,相同类型的细胞可以聚集到一起(图4B,第二三四列,分别为inClust在监督,半监督,无监督的结果)。在同类细胞形成的聚类中,包含了多个批次的样本(图4C,第二三四列)。而与现有同类主流68种算法的对比,可以看到inClust的总得分遥遥领先(图5)。
图5:inClust在标准数据集上与多种现有方法的对比。
一个细胞的表达谱受到多个协变量的影响,比如供体,以及是否受到刺激物刺激等等。不同协变量的组合会对细胞的表达谱造成不同的影响。如何根据已知的协变量以及协变量组合对细胞表达谱的影响来预测未知协变量组合对细胞表达谱的影响,可以定义为分布外样本生成问题(Out-Of-Distribution (OOD) generation)。
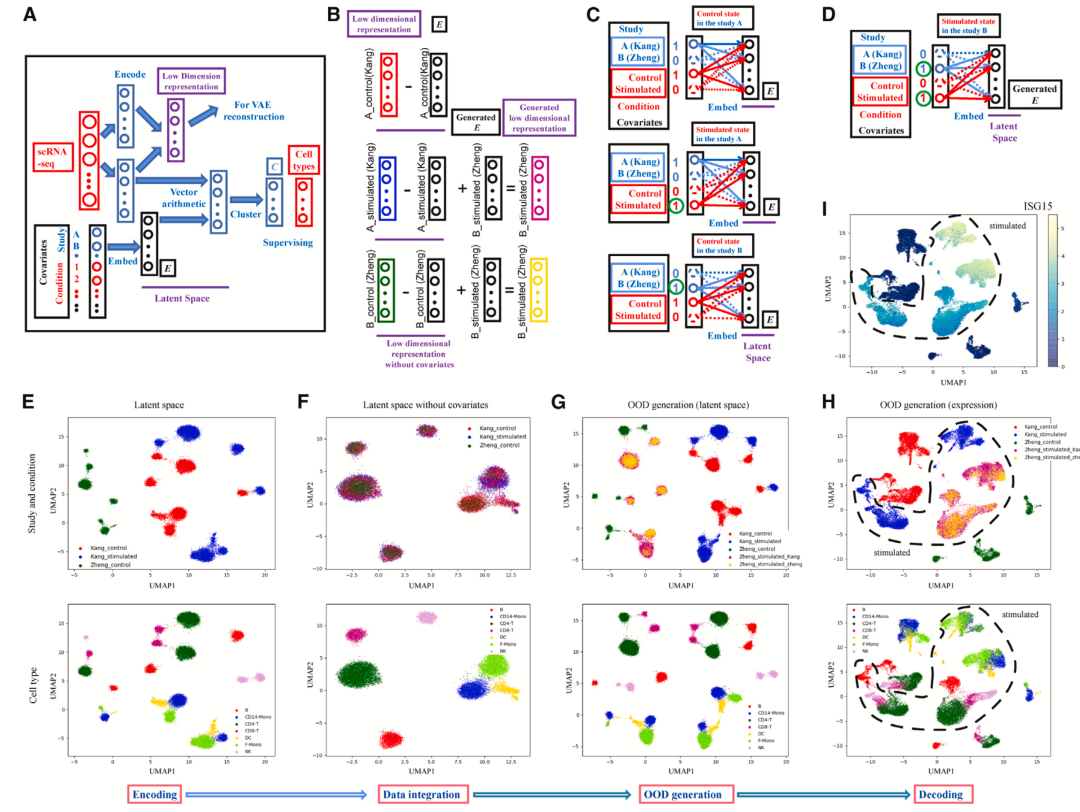
如下就是一个分布外样本生成问题的实例:已知三组样本,分别来自两个供体(图6E)。其中一个供体的样本包含正常和受刺激物刺激的样本(图6E,红色和蓝色),另一个供体仅包含正常的样本(图6E,绿色)。
这里的协变量就是:1 供体,2 是否受刺激,样本外生成问题就是要生成绿色样本供体受刺激物刺激后的结果。InClust 通过表达谱编码,协变量嵌入,低维空间向量运算,以及分类约束,能精确地在低维空间表征已知的协变量以及协变量组合(图6A,B,C),以及不受协变量影响的表达谱(图6B,F)。利用这些在低维空间中精确表征的协变量,inClust 能够生成新的协变量组合(图6D),并进一步在低维空间生成分布外的样本(图6B,G,紫色和金黄色样本为绿色样本受刺激后的产物)。该生成的分布外样本经过解码,回到原有的高维空间,其特征包括受刺激后的应激反应(图6H,I),证明了 inClust 能够较好地生成绿色样本受刺激后的结果,即分布外的样本。
图6:Inclust在有监督模式中,分布外样本生成(OOD)
单细胞转录组的自动标注不同于传统的有监督分类问题,其有两大难点:一个是批次效应(Batch effect),一个是部分重叠(Partial overlap)。批次效应是指参考集(带有标签的数据)与查询集(需标注的数据)之间有系统性的偏差,需要去除批次效应以后才能正常标注(图7A)。部分重叠是指参考集与查询集都有自己独有的细胞类型,标注方法需要找出查询集新的细胞类型(图7B)。InClust就是为了同时解决这两个问题。低维空间的向量运算能够将批次效应从转录组数据中去除(图7C)。
同时,将聚类类别数量设定为多于参考数据集类别数量。这样,聚类类别可以分为两类,半监督聚类类别(semi-supervised cluster)和无监督聚类类别(unsupervised cluster)。半监督聚类类别包括参考集数据和查询集数据,在该类别中,细胞类型标签从参考集转移到查询集。无监督聚类类别只包含查询集数据,可根据该类别高表达基因来进行标注(图7D)。
从结果来看,InClust解决了这两个问题:不同批次的样本聚到一起(图7E);半监督聚类类别完成自动标注(标签转移)(图7F),无监督聚类类别识别查询集特有细胞类型(图7G)。因此,InClust可以解决单细胞转录组的标注(图7H)。
图7:inClust在半监督模式下的标签转移和新样本识别
空间转录组包括转录组和空间位置两部分信息,在无监督模式下,inClust能够整合这两部分信息,将整个空间转录组划分为不同的空间域(spatial domain),更好地揭示转录组在空间上的异同(图8A)。相比图8D使用 k-means 聚类的结果,inClust的对空间域的划分边界更清晰(图8C),异常值(outlier)也更少(图8E)。
图8:InClust在无监督模式下,对小脑的空间转录组数据进行空间域的划分。
一个细胞的表达谱受到多个协变量的影响,或者是生物的(例如条件、刺激物),或者是非生物的(例如批量、技术)。不同的协变量组合通常导致不同的转录谱。随着技术的发展,产生了包含多种来源信息的复杂数据集,可以作为聚类的输入。传统的聚类方法只从一个来源获取信息(例如,基因表达)。因此,需要集成来自多个来源的信息进行聚类,并准确预测不同来源的协变量造成的影响。
相比其它方法,InClust 的优势在于其模块在实现上是独立的,但在运行时是相互关联的,可以同时完成数据整合与数据拆分 (data integration and decomposition)。嵌入模块可以将任意向量(协变量)嵌入到潜在空间中,从而不仅整合样本来源信息,还有潜力将来自不同组学的信息汇总,从而进行多组学分析。矢量算术模块可以通过减法去除不必要的协变量,也可以通过加法合并辅助信息,灵活地对信息进行集成。聚类模块不仅可以对包含多个来源信息的隐空间向量进行分类,还可以灵活地利用数据中的标签信息,使模型成为一个通用的深层生成框架,可以在从有监督、半监督以及无监督这三种模式下进行聚类。
在有监督模式下,inClust 能够将不同协变量对表达谱的影响逐一剥离,在低维空间精确地表征各种协变量和不受协变量影响的表达谱;在半监督模式下,inClust可以除去批次效应,并将查询集分解为参考集中出现过的类型和查询集独有的类型,解决部分重叠问题;在无监督模式下,inClust可以整合转录组数据和空间位置信息来划分空间域。因此,inClust可以很好地完成数据整合与数据拆分。
由于 inClust 中使用变分自编码器(VAE),使其不像基于自动编码器(AE)的模型那样容易受到过拟合风险的影响。inClust 被证明对数据缺失具有鲁棒性;额外的噪声不会影响 inClust 的性能。简而言之,inClust 是单细胞转录组学领域多任务协调和分解的理想框架。
王力飞,基因组学博士,毕业于中国科学院北京基因组研究所,现就职于浙江树人大学树兰国际医学院,集智科学家。
学者主页:https://pattern.swarma.org/user/324
大模型与生物医学:
AI + Science第二季读书会启动
生物医学是一个复杂且富有挑战性的领域,涉及到大量的数据处理、模式识别、理论模型建构和实验验证等问题。AI基础模型的引入,使得我们能够从前所未有的角度去观察和理解这个领域的问题,加速科学研究的步伐,提高医疗服务的效率和效果。这种交叉领域的合作,标志着我们正在向科技与生物医学深度融合的新时代迈进,对于推动科学研究、优化医疗服务、促进人类健康有着深远的影响。
集智俱乐部联合西湖大学助理教授吴泰霖、斯坦福大学计算机科学系博士后研究员王瀚宸、博士研究生黄柯鑫、黄倩,华盛顿大学博士研究生屠鑫明,共同发起以“大模型与生物医学”为主题的读书会,共学共研相关文献,探讨基础模型在生物医学等科学领域的应用、影响和展望。读书会从2023年8月20日开始,每周日早上 9:00-11:00 线上举行,持续时间预计8周。欢迎对探索这个激动人心的前沿领域有兴趣的朋友报名参与。
详情请见:
大模型与生物医学:AI + Science第二季读书会启动
点击“阅读原文”,报名读书会