AI模拟细胞:让活细胞在大数据中自发涌现!

导语
“我们能在计算机里构建一个细胞吗?” 这一根本性问题,正随着人工智能和组学技术的进步而迎来突破性机遇。最近,权威期刊Cell发表了一篇重磅综述,深入探讨了 “人工智能虚拟细胞”(AI Virtual Cell, AIVC) 这一前沿概念。这篇文章汇集了AI与生命科学领域的顶尖智慧。作者团队阵容非常强大,横跨学术界(如斯坦福、哈佛 、科技巨头(如谷歌研究院、微软研究院)和生物技术行业(如基因泰克),并由陈-扎克伯格计划 (CZI)的研究者联合推动,共同为构建“AI虚拟细胞”绘制蓝图。本文是对该综述的翻译。
细胞对于理解健康和疾病至关重要,但其作为生命最基本功能单位的复杂性,让传统建模方法在模拟其功能与行为时显得力不从心。如今,我们有机会创建一个人工智能虚拟细胞——一个基于大型神经网络的多尺度、多模态模型,能够表征和模拟分子、细胞乃至组织在不同状态下的行为。这种AIVC不仅是一种新的建模工具,更是一种全新的科学范式。
通过实现高保真度的仿真,AIVC有望让科学家在虚拟空间进行复杂的“计算机实验” (in silico experimentation),成为理解疾病发生、药物反应和细胞工程的强大工具,甚至将细胞重新定义为一种信息处理系统。然而,构建AIVC是一项巨大的挑战,需要整合跨尺度、跨物种的多样化数据,并建立一个开放协作的科学共同体。如同历史上的“基因组计划”(human genome project)一样,AIVC的提出标志着一个重要的历史关口,它不仅是一个学术愿景,更是一种面向未来的科学基础设施。
展望未来,虚拟细胞平台或许会像显微镜一样普及,帮助科学家高效地验证假设、设计实验。这篇综述所描绘的,正是一幅关于未来生物学研究如何被深刻重塑的宣言。其最终目标并非替代自然,而是为了与生命世界展开更科学、高效的对话。
关键词:细胞生物学、人工智能、机器学习、虚拟细胞
王昊丨作者
周莉|审校

论文题目:How to build the virtual cell with artificial intelligence: Priorities and opportunities
论文链接:https://www.cell.com/cell/fulltext/S0092-8674(24)01332-1
论文来源:Cell
-
摘要
-
引言
-
建立AIVC所面临的重大挑战
-
AIVCs的关键能力
-
构建AIVC的方法
-
构建AIVC的数据需求和要求
-
AIVC的模型评估和可解释性
-
前景展望
细胞对于理解健康和疾病至关重要,然而传统模型在建模和模拟细胞功能与行为方面存在不足。人工智能和组学技术的进步为创建 AI 虚拟细胞(AIVC)提供了突破性机遇,这是一个基于大型神经网络的多尺度、多模态模型 (a multi-scale, multi-modal large-neural-network-based model),能够表征和模拟分子、细胞和组织在不同状态下的行为。本文提出了 AIVC 设计的愿景,以及构建 AIVC 的协作努力将如何通过实现高保真度仿真、加速科学发现和指导实验研究来变革生物学研究,为理解细胞功能和促进开放科学中的跨学科合作提供新机遇。
1. 背景
1. 背景
细胞,作为生命的基本单位,是一个极其复杂的实体,其性质和行为挑战着物理和计算建模的极限。每个细胞都是一个动态的适应性系统,其复杂的行为从无数分子相互作用中涌现出来。某些方面对扰动具有显著的鲁棒性,比如基因的缺失或用来自不同物种的同源基因替换。而其他方面则对看似微小的干扰都很敏感,比如点突变或外部因子,这些都可能使细胞陷入功能失调和疾病状态。
为了理解细胞的功能,科学家们试图构建虚拟细胞模型(virtual cell models),以模拟、预测和调控细胞行为 [1–6]。现有的细胞模型通常基于规则(rule-based),将对潜在生物机制的假设与从观测数据中拟合的参数相结合。这些模型往往依赖于明确定义的数学或计算方法,例如微分方程 [7–9]、随机模拟 [10,11] 或基于智能体的模型 [12,13]。它们在复杂性上存在差异,涵盖细胞生物学中多个明确的研究方向,如转录与翻译 [14]、细胞骨架驱动的细胞行为 [15,16]、生化网络 [17] 以及代谢通量 [18,19]。首个全细胞模型于 2012 年开发,其代表了生殖支原体(Mycobacterium genitalium)这一生物体中已知的全部 482 个基因及分子功能 [8]。自这项开创性工作以来,研究者们已开发出全基因组模型,用以代表包括大肠杆菌在内的其他细菌生物体 [8,20–22]。
尽管这些方法在生物系统建模中广泛应用,但迄今为止的方法都无法充分捕捉细菌和更复杂系统(如人类细胞)运行的许多方面。 这些挑战共同构成了构建虚拟细胞的重大障碍,体现在多个层面。 首先,在多尺度建模 (Multi-scale modeling)方面,细胞在时间和空间上跨越从原子、分子到细胞和组织的多个尺度运行,其功能特性通过尺度间的非线性转换而产生 。其次,细胞功能包含众多相互作用且多样化的过程 (Diverse processes with massive numbers of interacting components),如基因调控、代谢和信号转导(transduction),每个过程都涉及大量处于动态构型中的生物分子种类 。最后,大多数细胞过程表现出高度的非线性动力学 (Nonlinear dynamics),输入的微小变化可能导致输出的复杂变化,这进一步增加了建模的难度 。
科学技术中的两次激动人心的革命——人工智能革命和组学革命——使得直接从数据中学习构建细胞模型成为可能。这两次并行的革命为一个雄心勃勃的愿景提供了前所未有的机遇,即构建 AI 虚拟细胞 (AIVC),这是一个基于大型神经网络的多尺度、多模态模型,能够表征和模拟分子、细胞和组织在不同状态下的行为(图 1)。
在实验方面,测量技术通量的指数级增长使得在不同细胞和组织系统内部及之间收集了大量且不断增长的参考数据集 [23–25],在过去几年中数据每 6 个月翻一番 [26],同时这些测量还能与系统性扰动相结合[27–29]。在计算方面,人工智能技术的快速发展增强了我们直接从数据中学习模式和过程的能力,而无需明确的规则或人工标注 [30,31]。这种建模范式已在生物分子领域得到成功应用,例如,从序列预测三维 (3D) 分子结构[32–34] 以及不同分子组分之间的相互作用 [35–38]。其中,Google DeepMind 的 AlphaFold 在该领域取得了突破性进展。人工智能领域最新的建模方法提供了表征和推理工具,满足了预测性 (predictive)、生成性 (generative) 和可查询性 (queryable)这三个关键特性,这些都是推进生物学研究和理解的关键功能。
基于这些特性,我们认为现在已经具备了开发完全数据驱动的基于神经网络的 AIVC 表征的方法,这可以通过实现快节奏的计算机模拟研究以及在计算方法和验证性湿实验室实验之间建立强大桥梁来加速生物医学研究(图 1)。
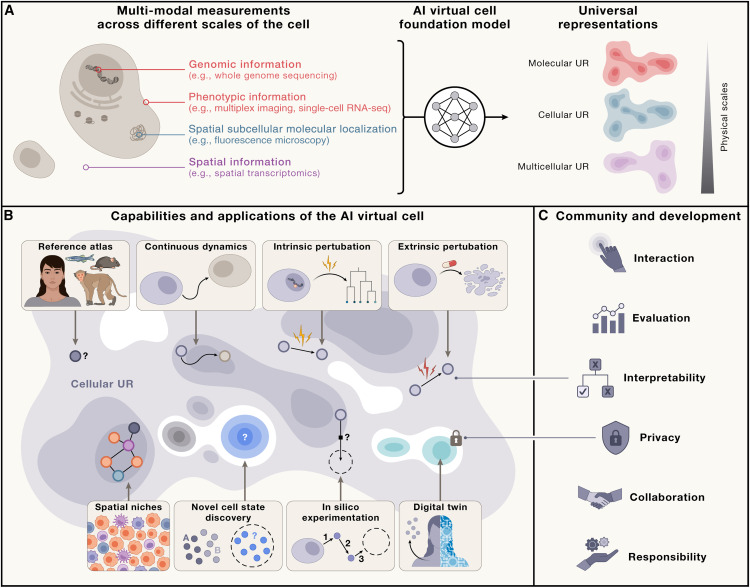
图 1. AIVC 的能力
(A) AIVC 提供了一种通用表示 (universal representation, UR),可以跨物种和条件获得细胞状态,并可从跨尺度 (分子、细胞和多细胞) 的不同数据模态中生成。
(B) AIVC 具备表示和预测细胞生物学特性的能力。这种通用性使该表示能够作为参考基准,泛化到此前未观察到的细胞状态,为未来的数据生成提供指导。由于该表示在不同模态间共享,其生成不受特定数据类型的限制,可作为跨模态统一分析的虚拟表示。AIVC 还能够对细胞在不同状态间转换的动力学过程进行建模,无论是分化等自然过程、遗传变异导致的转换,还是通过工程扰动人为引发的转换。因此,AIVC 能够实现计算机模拟实验,而这类实验在实验室中可能成本过高或根本无法开展。
(C)AI虚拟细胞(AIVC)的最终价值,取决于其在不同层面与人类的有效互动。对于个体科学家而言,其潜力必须通过开放的许可证和普惠的计算资源来充分释放,确保广泛的可及性 。同时,可以借助语言模型等中介层来增强其可解释性,让虚拟细胞能清晰地传达其复杂的分析结果。在更广泛的科学界层面,对AIVC的评估不应局限于狭隘的基准测试,而应聚焦于其核心能力的验证 。一个开放、活跃的社区生态对于模型的持续迭代、改进和保持开放性至关重要 。最终,在社会层面,AIVC必须将数据隐私和安全作为基石,通过强大的隐私保护措施来守护敏感信息,从而建立公众信任。
2. 建立AIVC所面临的重大挑战
2. 建立AIVC所面临的重大挑战
-
概述能力并设计评估框架
生物学领域中数量激增的基础模型 (foundation models) 执行着本文所概述的虚拟细胞能力的一个子集。鉴于这些方法的多样性,定义 AIVC 的核心能力应该是什么以及如何评估这些能力变得非常重要。对于每一种能力,都必须设计适当的指标,并收集全面的评估数据。模型的能力应该在一般性能以及回答特定生物学问题的能力方面进行评估。持续改进基准测试策略与 AIVC 模型同步发展,并确保它们与生物学上有意义的目标保持一致,这一点至关重要。随着该领域在这些问题上达成更好的共识,合作机会将会出现,生成虚拟细胞的速度也将加快。
-
在不同架构的变化情境中建立自一致性
生物学极其复杂:它在不同尺度、不同环境中运作,并通过不同模式进行测量。AIVC 模型必须在所有这些维度上保持自洽性 (self-consistent) 。 具体来说,这种自洽性体现在两个层面:跨物理尺度的一致性:模型的功能必须能在不同物理尺度间贯穿体现。例如,分子间的相互作用,无论是在测量其结合亲和力 (binding affinity),还是在观察其对基因表达、细胞间通讯或组织结构的影响时,都应产生逻辑一致且相互吻合的效应。以及跨数据模式的一致性:模型的预测结果应当独立于其输入和输出的数据模式。这意味着,同一个生物实体,即使用不同的技术进行分析,在AIVC中也应被赋予统一的内部表征。
-
平衡可解释性和生物学效用
深度学习方法在生物学中应用的一个一致趋势是,在大型基础模型兴起的推动下,模型性能提升与其日益不可解释的 “黑盒” 特性之间存在隐性权衡。AIVC 模型最终将根据其扩展我们对生物学理解的能力来评判,无论是通过为生物过程提供新颖见解,还是通过加速科学研究过程。
为实现这一目标,AIVC 模型必须做出高度准确且校准良好的预测来模拟生物学,并且必须平衡可操作性与可解释性之间的权衡。可操作的模型输出是那些对设计经济高效的验证实验具有高效用的输出,是初期实际应用的关键。解释模型预测存在多种方法,包括因果建模、稀疏特征化和反事实推理,这是一个高度活跃的研究领域。构建直观的界面,通过其他模型(如 AI 研究智能体)促进 AIVC 的研究和解释,将进一步提高下游效用。
-
构建协作细胞建模框架
AIVC 的成功开发需要跨学科合作。为此,我们展望未来的 AIVC 平台将发展成为一个开放、互联的综合性枢纽。一方面,它将作为科研中心,支持研究人员协作开发细胞模型并进行广泛部署;另一方面,它也将承担教育使命,为科研人员提供专业培训,并为教育工作者、患者及社会公众设计丰富的参与活动。因此,投资于促进 AIVC 开放协作开发的基础设施应当是高优先级事项。
-
确保 AIVC 使所有人受益并促进道德和负责任的使用
确保 AIVC 的开发和应用符合伦理标准,促进其负责任的使用至关重要。这包括制定明确的政策和指导方针,以确保所有相关方都能在透明和公正的环境中参与 AIVC 的研究和应用。此外,必须加强对 AIVC 技术的监管,以防止潜在的滥用和不当使用,确保其在科学研究和社会应用中的正当性和有效性。
-
生成反映人类多样性的大型开放数据集
生成反映人类多样性的大型开放数据集是训练 AIVC 模型不可或缺的,这也构成了一项重大挑战。开发者在构建AIVC时必须极其谨慎地确保这些数据集得到道德和透明的使用,并制定策略来降低模型被虚假数据污染的风险。从一开始就与伦理和监管专家密切合作,对于建立新的监管规范以促进 AIVC 的负责任使用至关重要。
-
了解不同数据类型的价值以优先考虑大规模数据生成
人工智能虚拟细胞协作开发的一个基本问题是应该收集哪些数据和模态,以实现跨生物学背景和尺度的泛化。这些数据需要涵盖不同物种、领域和模态的生物学广度,代表生命的异质性,同时保持足够的深度以区分真实信号和噪声。数据生成的一个关键方面是同时测量时间和物理尺度,同时允许对系统进行扰动。
3. AIVCs的关键能力
3. AIVCs的关键能力
我们对AIVC的构想是,它是一个能够学习并模拟细胞及细胞系统在多变条件与情境下行为的模拟器 (learned simulator) 。这些情境包括分化状态、扰动、疾病状态、随机波动和环境条件(图 1)。 它必须能够跨越不同的生物学尺度、时间维度和数据模式进行工作,并最终帮助揭示细胞系统的“编程语言”,同时为工程化应用提供一个可操作的接口。
具体而言,一个理想的AIVC需具备以下三项核心能力,以赋能研究人员:
(1) 创建通用表示 (Universal Representation, UR):能够跨越物种、数据模式、数据集和具体情境(包括细胞类型、发育阶段和外部条件),建立生物状态的通用表示。
(2) 预测功能与机制:能够预测细胞的功能、行为和动态变化,并揭示其背后的潜在机制。
(3) 执行计算机模拟实验 (In silico experiments):能够通过模拟实验来生成和检验新的科学假设,并指导后续的数据收集,从而高效地扩展虚拟细胞自身的能力 。
接下来,我们将对这些关键能力进行详细阐述。
-
通用表示 (Universal representation, UR)
AIVC 将生物数据映射到通用表征空间 (UR spaces)(图 1A),这不仅有助于洞察不同生物状态间的共享特征,还能作为一个全面的参照基准 。这些 UR 应能整合分子、细胞和多细胞这三个物理尺度,并容纳来自任何相关数据模式和背景的信息 。这种整合能力使研究人员可以在 AIVC 框架内,利用其丰富的存量生物学知识来补充新数据,从而弥合不同数据集之间的鸿沟。至关重要的是,这种多层次的表示应能泛化 (generalize) 到训练数据中未曾出现过的新状态。这种涌现能力 (emergent capability) 将解锁对全新生物学状态的发现,这些状态可能从未被直接观察到,甚至在自然界中本不存在 。例如,AIVC 通过学习巨噬细胞的炎症状态,或许能预测出小胶质细胞中一种前所未知的炎症状态。此外,AIVC 还应能预测由干预措施(或为达到特定状态所需的干预措施)所产生的新状态,这将为细胞工程和合成生物学领域的下游应用提供广阔的可能性。
-
预测细胞行为和理解机制 (Predicting cell behavior and understanding mechanisms)
AIVC的一项决定性功能 (defining function) 是其建模细胞响应和动力学的能力 。通过在覆盖不同情境和尺度的大量快照式、时间分辨、干预性和非干预性数据集上进行训练,AIVC 可以深入理解在自然或工程信号(如化学、遗传扰动及其组合)驱动下发生的分子、细胞和组织层面的动力学过程 。AIVC应能预测那些在实验室中从未测试过的扰动所产生的响应,并能考虑到扰动发生时特定细胞环境的特征。
同时,AIVC 也应能模拟细胞状态随时间的演变,以及由此产生的多细胞空间布局 。通过对细胞整体状态的瞬时特性和持续变化的建模,AIVC 可以揭示在发育、稳态维持、疾病发生与发展等多样化动态过程中,此前未被研究过的演化轨迹 。
另一个关键挑战是理解这些表型和轨迹背后的分子机制 。通过模拟不同干预措施的效果,AIVC 可以提出潜在的因果因素 。尽管仅凭计算不一定能完全揭示因果关系,但AIVC 有潜力缩小可能假设的空间 (reduce the space of possible hypotheses) 。它能提出与不同表型相关的潜在因果因子,并给出相应的不确定性程度,从而让科学家能更有针对性地通过实验进行验证 。
-
计算机模拟实验和指导数据生成
在现实应用中,AIVC 的另一项决定性功能是其指导数据生成和实验设计的能力 。研究人员应能通过当前实验室实验的计算孪生体 (computational twins)——在此我们称之为虚拟仪器 (virtual instruments, VIs)——来查询 AIVC 。例如,虚拟实验可以模拟那些难以在体外培养的细胞类型的实验,或者从低成本测量(如无标记成像)中推断出昂贵的读数(如单细胞转录组) 。虚拟实验还能以实验室无法企及的规模筛选海量的潜在扰动源,这在面对组合扰动的指数级搜索空间时,显得尤为宝贵。
AIVC 将引领一种设计生物实验的新范式 。在此范式中,AIVC不仅能设计实验来验证特定的科学假设,还能设计实验来增强自身的能力 。AIVC能为其预测分配置信度值,从而实现交互式查询,引导实验者为低置信度区域生成最有效的数据以进行模型优化 。通过扩展为一个主动、迭代的实验室在环”(lab-in-the-loop)流程, AIVC 的性能将得到高效且有针对性的提升 。最终,AIVC甚至可能能够识别出自身在生物学理解上的关键空白,并提出填补这些空白的最有效路径 。
4. 构建AIVC的方法
4. 构建AIVC的方法
我们设想 AIVC 是一个综合的 AI 框架,由几个相互连接的基础模型 (foundation models) 组成,这些模型在日益复杂的组织层次上表示动态生物系统——从分子到细胞、组织及更高层次。
我们的方法有两个主要组成部分:
-
通用多模态多尺度生物状态表征(UR) (a universal multi-modal multi-scale biological state representation)
-
一组 VI (virtual instruments, VIs) ——即操作或解码这些表征的神经网络。
尽管可能存在构建 AIVC 的其他方法,但我们相信这种方法将提供一个可以协作和开放方式的扩展支架。
4.1 通用表示(UR)
UR 是由 AIVC 基础模型产生的嵌入 (embedding),它将高维、多尺度、多模态的生物数据转换为能保留有意义关系和模式的数值表示。AIVC 通过表示以下三个不同的物理尺度 (physical scales) 来捕获细胞生物学:
分子尺度 (Molecular scale):单个细胞内发现的分子及其结构;
细胞尺度 (Cellular scale):作为这些相互作用分子和结构的空间集合的单个细胞;
组织尺度 (Multicellular scale):单个细胞如何在组织中彼此相互作用以及与非细胞环境相互作用。
这三个尺度中的每一个都由一个独特的 UR 表示,并建立在前一层生成的抽象之上,从而连接不同的尺度。
4.2 虚拟仪器(VI)
在 UR 的背景下,VI 是以 UR 为输入并产生期望输出的神经网络。我们描述了两种类型的 VI:
-
解码器 VI(Decoder VI)
-
输入:UR
-
输出:人类可理解的结果,例如细胞类型标签或合成显微镜图像。
-
操控器 VI(Manipulator VI)
-
输入:UR
-
输出:另一个 UR,例如扰动后改变的细胞状态。
由于这些 VI 在相同的表示上运行,它们可以在不同的用例、实验和数据集之间共享和复用。 因此,我们设想任何科学家都能够在 UR 的基础上构建 VI 并与社区共享。 此外,构建与真实仪器(如显微镜)高度相似的 VI,可能促进特定仪器的实验室闭环系统的发展。
4.3 跨物理尺度构建 UR
每个表征普遍适用于特定类别的生物实体。这种抽象使虚拟细胞能够在这个通用框架实现无缝演进以及整合新数据,无论是来自新模态还是来自分布外来源的数据。
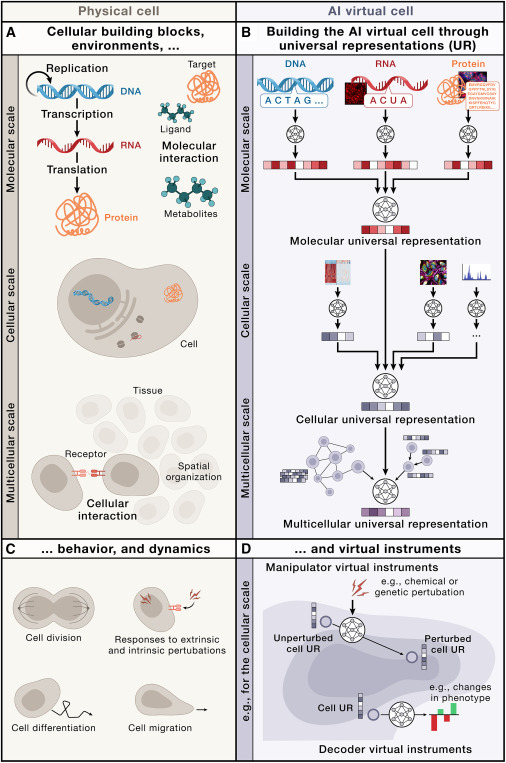
图2. AIVC 将是一个 多尺度基础模型,在每个物理尺度上学习生物实体的不同表征(图 2C)。这些表征可以聚合在一起并转换,以产生下一个更高物理尺度的表征。并形成一种递归架构模式,从单个分子水平应用到整个组织和器官尺度,使模型在不同生物尺度间保持一致性(图 2A)。
4.4 构建 AIVC 的人工智能方法
AIVC 将整合多种多样的神经网络架构。尽管这些架构并非专为生物学应用设计,但它们在与特定的生物模式和数据特性(inductive biases,归纳偏置)相匹配时,已展现出各自的优势。许多情况下,这些架构可以相互替代,因此需要根据它们在准确性、速度和通用性方面的表现进行权衡取舍。此外,研究社区正积极开发专门针对(大型)生物数据集特征量身定制的 AI 架构。
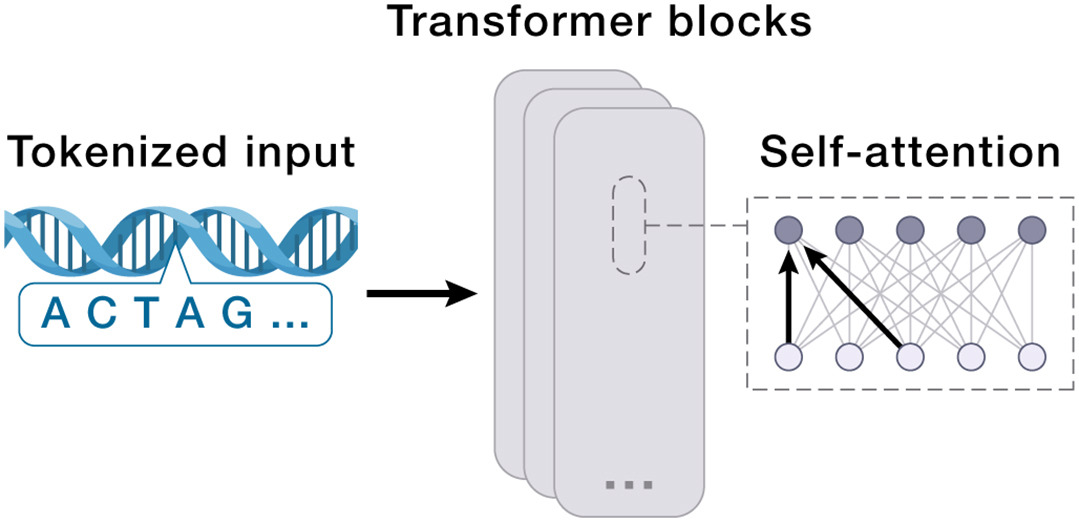
图3. 构建 AIVC 的人工智能方法之Transformer
-
Transformer神经网络 (Transformer neural network):Transformer 神经网络 [30] 由多个 Transformer 层组成,每个 Transformer 层都采用一系列标记(离散的信息片段,如单词、RNA 分子或基因表示)作为输入——第一层的初始标记和后续层的前一层输出。
在每一层中,标记使用自注意力来集成来自其他标记的上下文,增强自己的表示,然后通过前馈网络进行处理。这种架构从根本上只需要标记集合,可以很好地适应各种应用程序和用例。
默认情况下,传递给 Transformer 的标记集合没有任何排序。此外,自注意力机制是 Transformer 成功的核心,可以被视为一种强烈的生物归纳偏差。例如,在通过 scRNA-seq 检测到的 RNA 分子来表示细胞时,每个 RNA 分子(表示为标记)与其他 RNA 分子相互作用,通过自注意力模拟基因相互作用 [30]。使用基因的数字表示定制输入标记进一步允许集成从单个基因到整个细胞的不同生物数据尺度 [59, 60]。
此外,将位置编码引入标记使 Transformer 能够通过合并特定于序列的依赖关系来处理序列(例如自然语言 [30] 或生物序列如 DNA [48,142])。这种方法在掩码语言建模等应用中至关重要,其中模型预测序列中缺失的标记,从而增强其对数据中上下文关系的理解。
创新不断完善 Transformer,提高其处理更长序列的能力并提高效率。例如,状态空间模型等进步能够生成广泛的 DNA 序列 [51]。
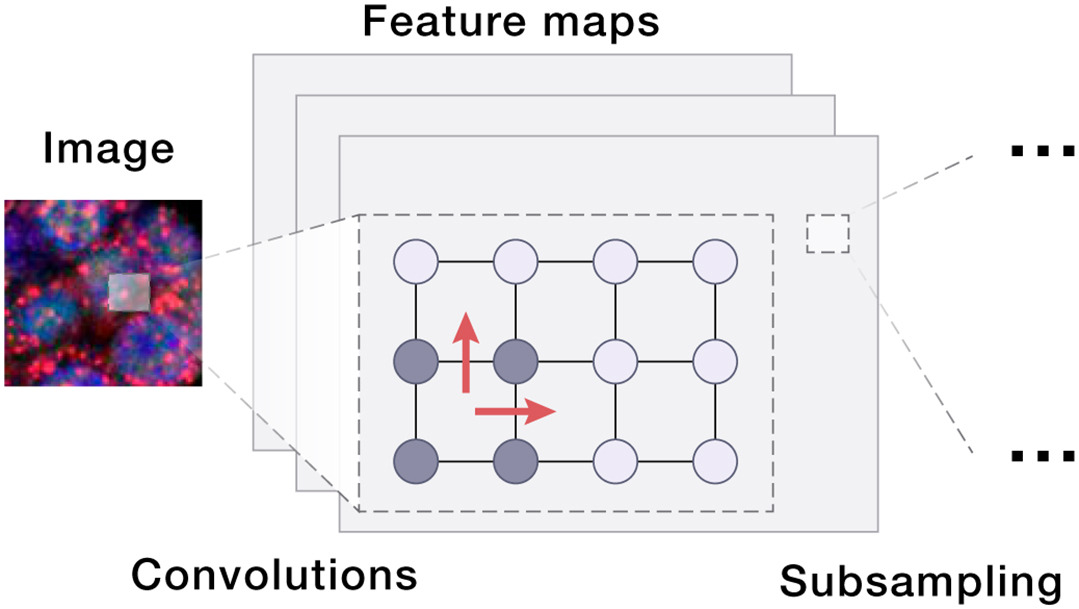
图4.构建 AIVC 的人工智能方法之CNN
-
卷积神经网络 (Convolutional Neural Network, CNN):
CNN 是一种深度学习模型,主要用于分析图像 [73,74]。它由多个层组成,通过反向传播自动自适应地学习特征的空间层次结构。卷积层、池化层和全连接层协同作用,从输入数据中提取特征并做出决策。
在生物学领域,CNN 已被证明对于涉及图像数据的任务具有无价的价值。例如:
-
细胞和组织的显微镜图像模式识别;
-
多重成像(同时标记和可视化多个目标) [143,144];
-
临床病理学中的 H&E 染色组织切片分析 [145];
-
活细胞成像 (live-cell imaging) 中用于跟踪细胞内或单分子随时间的动态变化 [146]。
图5.构建 AIVC 的人工智能方法之Diffusion模型
-
扩散模型 (Diffusion models):扩散模型是一类生成式深度学习模型,能够在多个领域生成高质量、多样化样本 [148]。其工作原理是:逐渐将随机噪声分布转化为结构化输出(图像、文本、细胞状态等)。
在扩散模型架构的基础上,flow matching 等方法可以模拟随时间演变的分布 [149],使其特别适合于动态变化和时间进展至关重要的生物学应用。
例如,它们能够生成细胞在时间和空间上的发育轨迹,或预测生物系统对治疗的反应 [87]。扩散与 flow matching 模型结合,为高维复杂生物数据的建模提供了强大工具。
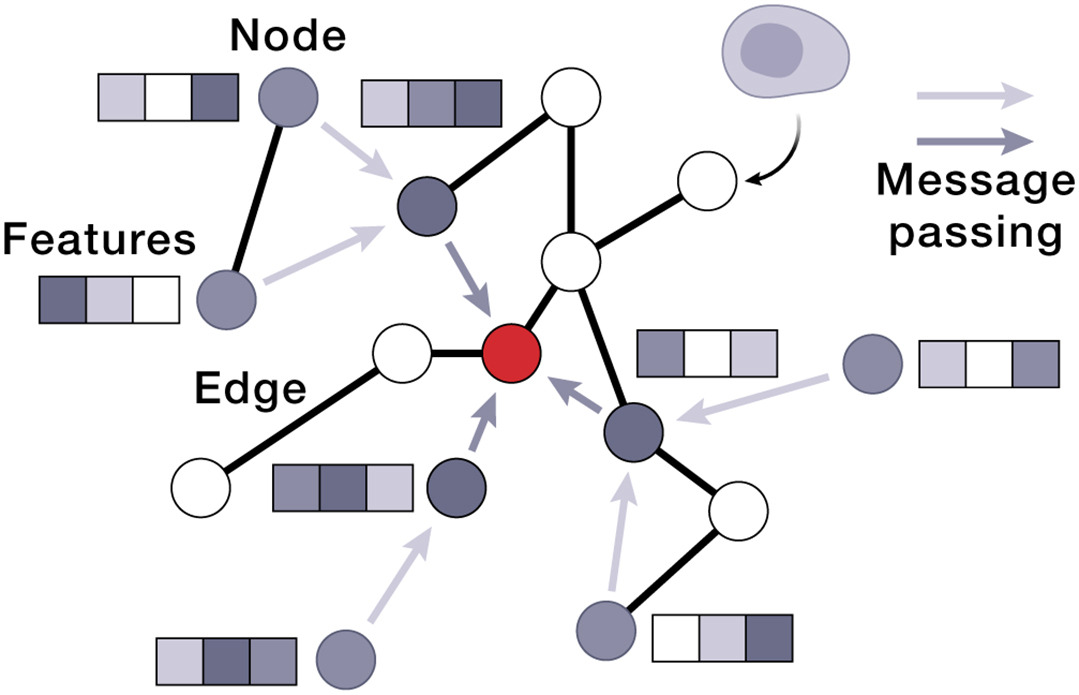
图6.构建 AIVC 的人工智能方法之GNN
-
图神经网络 (Graph Neural Network, GNN):是一类可以对图数据进行建模的架构 [150]。图由节点和边组成,是许多生物系统的自然表示:
-
蛋白质结构:残基作为节点,化学键作为边 [151];
-
组织结构:细胞作为节点,相邻细胞通过边表示物理或化学连接 [152, 153]。
GNN 可用于节点、边或整个图的预测 [154]。在节点级 GNN 中,每一层中节点会整合邻居的特征信息(消息传递机制),并逐层扩展感受野。在生物应用中,例如在空间转录组数据中,GNN 可以将每个细胞的基因表达作为初始特征,并在训练过程中通过邻居信息更新,从而揭示细胞间的空间相互作用与生态位 [153]。
5. 构建AIVC的数据需求和要求
5. 构建AIVC的数据需求和要求
AIVC 的一个关键考虑因素是必须收集哪些数据集和模态来实现其有效构建。与传统的实验设计不同,传统实验设计中数据是为了测试特定的科学假设而生成的,而用于训练 AIVC 的数据收集应该专注于确保 AIVC 所期望的广泛适用性和通用性。为了实现这些目标,数据理想情况下应该跨越不同的领域和模态,包含生物变异性的异质性和多样性,并使模型能够区分技术噪声、生物变异和生理差异。
数据生成需要同时探索时间和物理尺度,同时允许系统扰动。在这里,经典成像技术 [65, 100, 101] 包括活细胞成像,以及更新的结构成像技术,如冷冻电子断层扫描 (cryoelectron tomography) 和软X射线断层扫描 (soft X-ray tomography) [66, 102, 103],以及新颖的空间组学技术 (spatial omics technologies) [104, 105],为跨尺度建模生物分子和功能提供了机会。此外,生物过程跨越了巨大的时间尺度范围,从皮秒级的最快反应到持续数小时至一天的细胞分裂,再到历时数年的肿瘤发展,以及持续数十年的神经退行性变化。最近构建的通用细胞图谱 [101, 106] 可能成为建模较长时间尺度上细胞行为(如组织形成)的强大资源。需要新的方法来构建可比较的数据集,以捕获细胞在较短时间尺度上的行为,例如通过活细胞成像等方法。
除了分子测量外,数据收集的一个重要方面将是测量生物物理和生物化学细胞特性,为 AIVC 提供物理和化学现实主义的边界。
AIVC 发展的另一个重要驱动力将是多模态数据集 (multi-modal datasets) 。例如,连接分子和空间尺度的数据集,如单细胞转录组学数据结合组织学,以了解细胞如何相互作用以及哪些分子特征支撑着专门空间生态位的形成 [107]。需要进一步的技术发展来收集多模态数据,以捕捉分子特征、细胞行为、细胞调控和组织之间的关系。
尽管虚拟细胞建模的核心兴趣将集中在人类数据集上,目的是理解疾病并帮助开发新型治疗方法,但人类数据集在我们进行体内对照实验和扰动的能力方面是有限的。
在此,3D 组织生物学领域(包括类器官等培养系统)正逐渐兴起,成为一种在 3D 环境中研究复杂组织结构和功能[108] 的工具。它在 3D 环境中进行研究,同时允许对系统进行扰动。另一个超越这一限制的关键途径将是对演化历史的物种进行多样化的全机体谱分析,涵盖各种扰动和不同条件 [109, 110, 111]。理想情况下,大型数据集可以在所有三个物理尺度上被收集,这将使 AIVC 的应用能够从疾病研究扩展到工业生物技术、农业生物技术、传染病和气候变化等更广阔的领域。然而,基于目前在细胞和多细胞尺度上的数据收集趋势,对动物细胞进行建模仍然是最现实的选择。
最后,生物数据生成的一个关键方面将是对组合空间 (combinatorial spaces)的探索:生物空间通常是高维的,枚举其变体在一般情况下是难以处理的,例如,当考虑基因组的所有可能变体时。即使是少数实体的组合,如枚举扰动的成对或成组情况 [47, 90],实验设计也变得极其具有挑战性。鉴于组合的可能性会迅速膨胀,远超实验或计算的可行范围,因此必须开发探索这些可能性新方法。
-
构建 AIVC 需要多少数据?
原始生物数据的规模是不可否认的,但即使是一个人体细胞系统的纯粹标称规模也同样庞大,这使得第一性原理估算变得困难。例如,生物序列数据的短读长存档拥有超过 14 petabytes的信息 [112],这比训练 ChatGPT 所用的数据集大 1000 多倍 [113]。如果用于训练,这些数据的很大一部分可能是冗余的或具有递减收益,因此必须彻底研究模型性能的规模法则。
除了数据规模外,数据的多样性和质量对确保模型性能至关重要 [114]。来自人类和模式生物(如小鼠和大肠杆菌)的数据在序列和文献数据库中的代表性不均等,当用于训练时,会编码强烈的物种偏差 [114]。其他偏差,例如在性别、特定疾病或人类祖先群体方面的偏差,也可能降低 AIVC 模型的影响力 [115]。
尽管数据方面需要付出努力,但驱动 AIVC 的 AI 模型必须设计得能够承受并适应这些挑战,即在整合各种来源和质量的数据集的能力方面表现出稳健性。考虑到实验室技术的快速发展(这使得无法在单一平台上实现标准化)以及 AIVC 必须涵盖的模式和细胞系统的广泛多样性,这一点至关重要。随着虚拟细胞工作的成熟,开发模型的科学家、生成实验数据的科学家和资助机构之间的对话必须进一步加强。
6. AIVC的模型评估和可解释性
6. AIVC的模型评估和可解释性
对于 AIVC 发展而言,更重要的问题可能不是 “我们如何构建它们?” 而是 “我们如何建立对其能力和准确性的信任?” 为此,需要一个全面且适应性强的基准测试框架(benchmarking framework)。AIVC 需要在众多生物学背任务中展现通用性 (generalizability) 。它必须考虑由于环境变化、感染、遗传变异以及其他引起分布偏移 (distribution shifts) 的因素而不断演变的动态分布。 [116]。
即使在可推广性之外,新兴能力(比如那些与大语言模型 (Large Language Models, LLMs) 相关的能力)也可能使 AIVC 模型能够外推到真正的分布外数据。在生物学背景下,在评估过程中可能很难确定这个边界是如何定义的。新分子、新细胞状态,甚至新物种都可能被认为在训练分布内。因此,对新数据的外推可能仅限于考虑设计自然界中不存在的生物实体。
这种评估类型已经在分子设计领域得到了考虑,因为语言模型创建的蛋白质,如 esmGFP [52] 或 OpenCrispr1 [53],突出了它们与任何天然存在的对应物之间的差异。如果在设计这些模型时以外推为目标,那么可能需要使用生物力学、基于物理或机制建模的额外归纳偏置、微调或偏好优化。
对 AIVC 的评估应同时优先考虑其泛化能力和发现新生物学规律的能力。泛化能力衡量了模型在未曾见过的语境中的表现,例如面对新型细胞类型或不同的遗传背景。可以通过跨模态重构任务来评估这种能力,比如根据先前未见过的细胞形态来预测基因表达,或是预测细胞状态显微图像序列中的下一张图像。评估泛化能力有助于建立人们对 AIVC 模型的信心,相信它能够捕捉核心生物学过程,并理解这些过程在不同情境下的变化。建立连接生物学尺度与模态的跨模态基准,对科研界而言是一项当务之急,因为这些任务不仅在生物学上具有实用价值,而且定义明确。
最终,评判 AIVC 模型应基于其能否开辟理解生物学的新途径。这样的评估标准能够确保模型开发与生物学相关性保持一致。最初最有价值的成果,可能在于生成有意义且可验证的实验假设。为此,与实验可验证表型相关的验证数据集可能很适用,例如细胞的生长速率、分子谱、被破坏的蛋白质-蛋白质相互作用,或转录因子结合。
随着 AIVC 能力的提升,我们必须考虑单纯的统计性能指标是否足够,或者可解释性 (interpretability)和生物学因果关系 (biological causality)是否会成为其核心要求。
-
可解释性和相互作用
在生物学中,科学发现的标志之一是为观察到的现象创建机制模型 (mechanistic models) 。在创建虚拟细胞时,我们可能不得不放弃构建完全机制模型的能力,转而学习能够从数据中泛化并预测超出观察范围的相互作用。然而,努力提高可解释性仍然是可取的。
每个 AIVC 预测都可以通过决定结果状态的相应多尺度相互作用来证实。例如,了解细胞亚系统或蛋白质复合物在疾病组织中如何被破坏,可以帮助开发治疗干预措施 [118,119]。AIVC 的模块化结构将使研究人员能够精确定位参与每个预测行为的特定基因、蛋白质或分子过程。
大型模型布线中的模式也可以被利用来揭示组合生物学相互作用,例如蛋白质之间的相互作用,这些相互作用可以投射到可解释的空间中,而不限制原始模型的通用性。尽管 AIVC 的许多能力依赖于预测任务,但生成机制假设可以为理解和进一步探索 AIVC 的预测提供实验途径,这对于 AIVC 的采用和使用至关重要。
最终,为 AIVC 构建一个交互层 (interactive layer) 将是关键所在,这将使不同专业水平的研究人员能够有效地理解和利用其预测结果。使用 LLMs 构建的AI智能体 (AI agents) 可以作为虚拟研究助手,为非专业人员提供直观的界面 [46,120]。利用它们对科学文献的广泛知识,这些语言模型可以为 AIVC 做出的预测提供更深入的见解。
7. 前景展望
7. 前景展望
遗传学和基因组学界已经创建了大型参考数据集,如人类基因组计划 (human genome project) [23]、HapMap [122]、癌症基因组图谱 (the Cancer Genome Atlas, TCGA) [123]、ENCODE [124]、基因型-组织表达 (the Genotype-Tissue Expression, GTEx) 项目 [125]、人类蛋白质图谱 (the Human Protein Atlas, HPA) [64,126]、人类细胞图谱 (the Human Cell Atlas, HCA) [24],以及越来越多的深度表型化、人群规模的生物样本库工作 [127]。得益于这些项目,现在有大量参考数据可用于训练机器学习模型。尽管这些努力将继续发展,但它们也催化了一项新的并行工作:创建细胞生物学的虚拟仿真,这是一种新的科学探究过程。
因此,AIVC 有潜力彻底改变科学研究过程,在生物医学研究、个性化医疗、药物发现、细胞工程和可编程生物学 (programmable biology) 领域带来未来的突破。作为虚拟实验室 (virtual laboratory),AIVC 可以促进计算机模拟实验数据与物理实验室结果之间的无缝对接。因此,我们期望 AIVC 能够为生物过程提供更统一的视角,促进科学家们在生物学涌现特性如何产生这一问题上达成共识。
通过连接计算机系统、现代生成式 AI 和 AI 智能体以及生物学的世界,AIVC 最终能够让科学家将细胞理解为信息处理系统,并构建生命的虚拟描述。随着 AIVC 扩展对细胞和分子系统的理解,它也将越来越多地允许我们对它们进行编程并设计新颖的合成系统。AI 模型已经被用于设计新的 CRISPR 酶 [53]、功能蛋白 [128],甚至整个原核生物基因组 [51]。细胞和基因组工程工具精度的快速进步将加速这一转变,AIVC 的不同实例化将在工程新的功能性生物学能力方面展开竞争,就像在表示和模拟生物学方面的竞争一样激烈。
参考文献
-
Slepchenko, B.M., Schaff, J.C., Macara, I., and Loew, L.M. (2003). Quan-titative cell biology with the Virtual Cell. Trends Cell Biol. 13, 570–576.
-
Johnson, G.T., Agmon, E., Akamatsu, M., Lundberg, E., Lyons, B.,Ouyang, W., Quintero-Carmona, O.A., Riel-Mehan, M., Rafelski, S.,and Horwitz, R. (2023). Building the next generation of virtual cells to un-derstand cellular biology. Biophys. J. 122, 3560–3569.
-
Marx, V. (2023). How to build a virtual embryo. Nat. Methods 20,1838–1843.
-
Goldberg, A.P., Szigeti, B., Chew, Y.H., Sekar, J.A., Roth, Y.D., and Karr,J.R. (2018). Emerging whole-cell modeling principles and methods. Curr.Opin. Biotechnol. 51, 97–102.
-
Georgouli, K., Yeom, J.-S., Blake, R.C., and Navid, A. (2023). Multi-scalemodels of whole cells: progress and challenges. Front. Cell Dev. Biol. 11,1260507.
-
Marucci, L., Barberis, M., Karr, J., Ray, O., Race, P.R., de Souza Andrade,M., Grierson, C., Hoffmann, S.A., Landon, S., Rech, E., et al. (2020). Com-puter-aided whole-cell design: Taking a holistic approach by integratingsynthetic with systems biology. Front. Bioeng. Biotechnol. 8, 942.
-
Lauffenburger, D.A., and Linderman, J.J. (1996). Receptors: models forbinding, trafficking, and signaling (Oxford University Press).
-
Karr, J.R., Sanghvi, J.C., Macklin, D.N., Gutschow, M.V., Jacobs, J.M.,Bolival, B., Assad-Garcia, N., Glass, J.I., and Covert, M.W. (2012). Awhole-cell computational model predicts phenotype from genotype.Cell 150, 389–401.
-
Mangan, S., and Alon, U. (2003). Structure and function of the feed-for-ward loop network motif. Proc. Natl. Acad. Sci. USA 100, 11980–11985.
-
Zopf, C.J., Quinn, K., Zeidman, J., and Maheshri, N. (2013). Cell-cycledependence of transcription dominates noise in gene expression.PLoS Comput. Biol. 9, e1003161.
-
Eling, N., Morgan, M.D., and Marioni, J.C. (2019). Challenges inmeasuring and understanding biological noise. Nat. Rev. Genet. 20,536–548.
-
Hellweger, F.L., Clegg, R.J., Clark, J.R., Plugge, C.M., and Kreft, J.-U.(2016). Advancing microbial sciences by individual-based modelling.Nat. Rev. Microbiol. 14, 461–471.
-
Gorochowski, T.E. (2016). Agent-based modelling in synthetic biology.Essays Biochem. 60, 325–336.
-
Thiele, I., Jamshidi, N., Fleming, R.M., and Palsson, B. (2009). Genome-scale reconstruction of Escherichia coli’s transcriptional and translationalmachinery: a knowledge base, its mathematical formulation, and its func-tional characterization. PLoS Comput. Biol. 5, e1000312.
-
Odell, G.M., and Foe, V.E. (2008). An agent-based model contrastsopposite effects of dynamic and stable microtubules on cleavage furrowpositioning. J. Cell Biol. 183, 471–483.
-
Popov, K., Komianos, J., and Papoian, G.A. (2016). MEDYAN: mechano-chemical simulations of contraction and polarity alignment in actomyosinnetworks. PLoS Comput. Biol. 12, e1004877.
-
Burke, P.E.P., Campos, C.B.L., Costa, L.D.F., and Quiles, M.G. (2020).M. G. A biochemical network modeling of a whole-cell. Sci. Rep.10, 13303.
-
Li, G., Liu, L., Du, W., and Cao, H. (2023). Local flux coordination andglobal gene expression regulation in metabolic modeling. Nat. Commun.14, 5700.
-
Fang, X., Lloyd, C.J., and Palsson, B.O. (2020). Reconstructing organ-isms in silico: genome-scale models and their emerging applications.Nat. Rev. Microbiol. 18, 731–743.
-
Stevens, J.A., Gru¨ newald, F., van Tilburg, P.A.M., Ko¨ nig, M., Gilbert,B.R., Brier, T.A., Thornburg, Z.R., Luthey-Schulten, Z., and Marrink,S.J. (2023). Molecular dynamics simulation of an entire cell. Front.Chem. 11, 1106495.
-
Maritan, M., Autin, L., Karr, J., Covert, M.W., Olson, A.J., and Goodsell,D.S. (2022). Building structural models of a whole mycoplasma cell.J. Mol. Biol. 434, 167351.
-
Ahn-Horst, T.A., Mille, L.S., Sun, G., Morrison, J.H., and Covert, M.W.(2022). An expanded whole-cell model of E. coli links cellular physiologywith mechanisms of growth rate control. npj Syst. Biol. Appl. 8, 30.
-
Venter, J.C., Adams, M.D., Myers, E.W., Li, P.W., Mural, R.J., Sutton,G.G., Smith, H.O., Yandell, M., Evans, C.A., Holt, R.A., et al. (2001).The sequence of the human genome. Science 291, 1304–1351.
-
Regev, A., Teichmann, S.A., Lander, E.S., Amit, I., Benoist, C., Birney, E.,Bodenmiller, B., Campbell, P., Carninci, P., Clatworthy, M., et al. (2017).The human cell atlas. eLife 6, e27041.
-
CZI Single-Cell Biology Program, Abdulla, S., Aevermann, B., Assis, P.,Badajoz, S., Bell, S.M., Bezzi, E., Batuhan, C., Jim, C., Chambers, S.,et al. (2023). CZ CELL3GENE discover: A single- cell data platform forscalable exploration, analysis and modeling of aggregated data. Preprintat bioRxiv.
-
Heimberg, G., Kuo, T., DePianto, D., Heigl, T., Nathaniel, D., Salem, O.,Scalia, G., Biancalani, T., Turley, S., Rock, J., et al. (2023). Scalablequerying of human cell atlases via a foundational model reveals com-monalities across fibrosis-associated macrophages. Preprint at bioRxiv.
-
Dixit, A., Parnas, O., Li, B., Chen, J., Fulco, C.P., Jerby-Arnon, L., Marja-novic, N.D., Dionne, D., Burks, T., Raychowdhury, R., et al. (2016). Per-turb-Seq: dissecting molecular circuits with scalable single-cell RNAprofiling of pooled genetic screens. Cell 167, 1853–1866.e17.
-
Srivatsan, S.R., McFaline-Figueroa, J.L., Ramani, V., Saunders, L., Cao,J., Packer, J., Pliner, H.A., Jackson, D.L., Daza, R.M., Christiansen, L.,et al. (2020). Massively multiplex chemical transcriptomics at single-cell resolution. Science 367, 45–51.
-
Feldman, D., Funk, L., Le, A., Carlson, R.J., Leiken, M.D., Tsai, F., Soong,B., Singh, A., and Blainey, P.C. (2022). Pooled genetic perturbationscreens with image-based phenotypes. Nat. Protoc. 17, 476–512.
-
Vaswani, A., Shazeer, N., Parmar, N., Uszkoreit, J., Jones, L., Gomez,A.N., Kaiser, L., and Polosukhin, I. (2017). Attention is all you need.Adv. Neural Inf. Process. Syst. 30.
-
Rombach, R., Blattmann, A., Lorenz, D., Esser, P., and Ommer, B. High- resolution image synthesis with latent diffusion models. In IEEE Confer- ence on Computer Vision and Pattern Recognition (CVPR), 10684–10695.
-
Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger,O., Tunyasuvunakool, K., Bates, R., Zˇ´ıdek, A., Potapenko, A., et al.(2021). Highly accurate protein structure prediction with AlphaFold. Na-ture 596, 583–589.
-
Baek, M., DiMaio, F., Anishchenko, I., Dauparas, J., Ovchinnikov, S., Lee,G.R., Wang, J., Cong, Q., Kinch, L.N., Schaeffer, R.D., et al. (2021). Ac-curate prediction of protein structures and interactions using a three-track neural network. Science 373, 871–876.
-
Lin, Z., Akin, H., Rao, R., Hie, B., Zhu, Z., Lu, W., Smetanin, N., Verkuil, R.,Kabeli, O., Shmueli, Y., et al. (2023). Evolutionary-scale prediction ofatomic-level protein structure with a language model. Science 379,1123–1130.
-
Gomes, J., Ramsundar, B., Feinberg, E.N., and Pande, V.S. (2017).Atomic convolutional networks for predicting protein-ligand binding af-finity. Preprint at arXiv.
-
Cunningham, J.M., Koytiger, G., Sorger, P.K., and AlQuraishi, M. (2020).Biophysical prediction of protein–peptide interactions and signaling net-works using machine learning. Nat. Methods 17, 175–183.
-
Torng, W., and Altman, R.B. (2019). High precision protein functional sitedetection using 3D convolutional neural networks. Bioinformatics 35,1503–1512.
-
Corso G., Sta¨ rk H., Jing B., Barzilay R., and Jaakkola T. (2023). DiffDock: Diffusion Steps, Twists, and Turns for Molecular Docking The Eleventh International Conference on Learning Representations.
-
Kudo, T., Meireles, A.M., Moncada, R., Chen, Y., Wu, P., Gould, J., Hu,X., Kornfeld, O., Jesudason, R., Foo, C., et al. (2024). Multiplexed, im-age-based pooled screens in primary cells and tissues with perturbview.Nat. Biotechnol., 1–10.
-
Roohani, Y., Huang, K., and Leskovec, J. (2023). Predicting transcrip-tional outcomes of novel multigene perturbations with GEARS. Nat. Bio-technol. 42, 927–935.
-
Bunne, C., Stark, S.G., Gut, G., Del Castillo, J.S., Levesque, M., Leh-mann, K.-V., Pelkmans, L., Krause, A., and Ratsch, G. (2023). Learningsingle-cell perturbation responses using neural optimal transport. Nat.Methods 20, 1759–1768.
-
Lotfollahi, M., Klimovskaia Susmelj, A., De Donno, C., Hetzel, L., Ji, Y.,Ibarra, I.L., Srivatsan, S.R., Naghipourfar, M., Daza, R.M., Martin, B.,et al. (2023). Predicting cellular responses to complex perturbations inhigh-throughput screens. Mol. Syst. Biol. 19, e11517.
-
Bunne, C., Krause, A., and Cuturi, M. (2022). Supervised Training of Con-ditional Monge Maps. Adv. Neural Inf. Process. Syst. 35, 6859–6872.
-
Bereket, M., and Karaletsos, T. (2024). Modelling Cellular Perturbationswith the Sparse Additive Mechanism Shift Variational Autoencoder.Adv. Neural Inf. Process. Syst. 36.
-
Huang, K., Huang, K., Lopez, R., Hutter, J.-C., Kudo, T., Rios, A., and Re-gev, A. (2023). Sequential Optimal Experimental De- sign of PerturbationScreens Guided by Multi-modal Priors. In International Conference onResearch in Computational Molecular Biology (Springer Nature),
-
Roohani, Y.H., Vora, J., Huang, Q., Liang, P., and Leskovec, J. (2024).BioDiscoveryAgent: An AI Agent for Designing Genetic Perturbation Ex-periments Preprint at arXiv.
-
Cleary, B., and Regev, A. (2024). The necessity and power of random,undersampled experiments in biology. Preprint at arXiv.
-
Ji, Y., Zhou, Z., Liu, H., and Davuluri, R.V. (2021). DNABERT: pre-trainedBidirectional Encoder Representations from Transformers model forDNA-language in genome. Bioinformatics 37, 2112–2120.
-
Brandes, N., Ofer, D., Peleg, Y., Rappoport, N., and Linial, M. (2022). Pro-teinBERT: a universal deep-learning model of pro- tein sequence andfunction. Bioinformatics 38, 2102–2110.
-
Celaj, A., Gao, A.J., Lau, T.T.Y., Holgersen, E.M., Lo, A., Lodaya, V., Cole,C.B., Denroche, R.E., Spickett, C., Wagih, O., et al. (2023). An RNA foun-dation model enables discovery of disease mechanisms and candidatetherapeutics. Preprint at bioRxiv.
-
Nguyen, E., Poli, M., Durrant, M.G., Kang, B., Katrekar, D., Li, D.B., Bar-tie, L.J., Thomas, A.W., King, S.H., Brixi, G., et al. (2024). Sequencemodeling and design from molecular to genome scale with Evo. Science386, eado9336.
-
Hayes, T., Rao, R., Akin, H., Sofroniew, N.J., Oktay, D., Lin, Z., Verkuil, R.,Tran, V.Q., Deaton, J., Wiggert, M., et al. (2024). Simulating 500 millionyears of evolution with a language model. Preprint at bioRxiv.
-
Ruffolo, J.A., Nayfach, S., Gallagher, J., Bhatnagar, A., Beazer, J., Hus-sain, R., Russ, J., Yip, J., Hill, E., Pacesa, M., et al. (2024). Design of highlyfunctional genome editors by modeling the universe of CRISPR-cas se-quences. Preprint at bioRxiv.
-
Peng, Z., Schussheim, B., and Chatterjee, P. (2024). PTM-mamba: aPTM-aware protein language model with bidirectional gated mambablocks. Preprint at bioRxiv.
-
Dai, B., Mattox, D.E., and Bailey-Kellogg, C. (2021). Atten- tion please:modeling global and local context in glycan structure-function relation-ships. Preprint at bioRxiv.
-
Yu, T., Yao, T., Sun, Z., Shi, F., Zhang, L., Lyu, K., Xuan, B., Liu, A., Zhang,X., Zou, J., et al. (2024). LipidBERT: A Lipid Language Model Pre- trainedon METiS de novo Lipid Library. Preprint at arXiv.
-
Krishna, R., Wang, J., Ahern, W., Sturmfels, P., Venkatesh, P., Kalvet, I.,Lee, G.R., Morey-Burrows, F.S., Anishchenko, I., Humphreys, I.R., et al.(2024). Generalized biomolecular modeling and design withRoseTTAFold All-Atom. Science 384, eadl2528.
-
Rosen, Y., Brbic´, M., Roohani, Y., Swanson, K., Li, Z., and Leskovec, J.(2024). Toward universal cell embeddings: integrating single-cell RNA-seq datasets across species with Saturn. Nat. Methods 21, 1492–1500.
-
Rosen, Y., Roohani, Y., Agrawal, A., Samotorcˇan, L., Tabula SapiensConsortium, Quake, S.R., and Leskovec, J. (2023). Universal cell embed-dings: A foundation model for cell biology. Preprint at bioRxiv.
-
Chen, Y., and Zou, J. (2024). GenePT: A Simple but Effective Foun- da-tion Model for Genes and Cells Built from ChatGPT. Preprint at bioRxiv.
-
Mahdessian, D., Cesnik, A.J., Gnann, C., Danielsson, F., Stenstro¨ m, L.,Arif, M., Zhang, C., Le, T., Johansson, F., Schutten, R., et al. (2021).Spatiotemporal dissection of the cell cycle with single-cell proteogenom-ics. Nature 590, 649–654.
-
Chandrasekaran, S.N., Cimini, B.A., Goodale, A., Miller, L., Kost-Ali-mova, M., Jamali, N., Doench, J.G., Fritchman, B., Skepner, A., Melan-son, M., et al. (2024). Three million images and mor- phological profilesof cells treated with matched chemical and genetic perturbations. Nat.Methods 21, 1114–1121.
-
Carlson, R.J., Leiken, M.D., Guna, A., Hacohen, N., and Blainey, P.C.(2023). A genome-wide optical pooled screen reveals regulators ofcellular antiviral responses. Proc. Natl. Acad. Sci. USA 120,e2210623120.
-
Thul, P.J., A˚ kesson, L., Wiking, M., Mahdessian, D., Geladaki, A., Ait Blal,H., Alm, T., Asplund, A., Bjo¨ rk, L., Breckels, L.M., et al. (2017). A subcel-lular map of the human proteome. Science 356, eaal3321.
-
McDole, K., Guignard, L., Amat, F., Berger, A., Malandain, G., Royer,L.A., Turaga, S.C., Branson, K., and Keller, P.J. (2018). In toto imagingand reconstruction of post-implantation mouse development at the sin-gle-cell level. Cell 175, 859–876.e33.
-
Nogales, E., and Mahamid, J. (2024). Bridging structural and cell biologywith cryo-electron microscopy. Nature 628, 47–56.
-
Bauda, E., Gallet, B., Moravcova, J., Effantin, G., Chan, H., Novacek, J.,Jouneau, P.H., Rodrigues, C.D.A., Schoehn, G., Moriscot, C., et al.(2024). Ultrastructure of macromolecular assemblies contributing to bac-terial spore resistance revealed by in situ cryo-electron tomography. Nat.Commun. 15, 1376.
-
Lelek, M., Gyparaki, M.T., Beliu, G., Schueder, F., Griffie´ , J., Manley, S.,Jungmann, R., Sauer, M., Lakadamyali, M., and Zimmer, C. (2021). Sin-gle-molecule localization microscopy. Nat. Rev. Methods Primers 1, 39.
-
Mo¨ ckl, L., and Moerner, W.E. (2020). Super-resolution microscopy withsingle molecules in biology and beyond–essentials, current trends, andfuture challenges. J. Am. Chem. Soc. 142, 17828–17844.
-
Cesnik, A., Schaffer, L.V., Gaur, I., Jain, M., Ideker, T., and Lundberg, E.(2024). Mapping the multiscale proteomic Or- ganization of cellular andDisease Phenotypes. Annu. Rev. Biomed. Data Sci. 7, 369–389.
-
Qin, Y., Huttlin, E.L., Winsnes, C.F., Gosztyla, M.L., Wacheul, L., Kelly,M.R., Blue, S.M., Zheng, F., Chen, M., Schaffer, L.V., et al. (2021). Amulti-scale map of cell structure fusing protein images and interactions.Nature 600, 536–542.
-
Dosovitskiy, A. (2020). An image is worth 16×16 words: transformers forimage recognition at scale. Preprint at arXiv.
-
Fukushima, K. (1980). Neocognitron: a self organizing neural networkmodel for a mechanism of pattern recognition unaffected by shift in po-sition. Biol. Cybern. 36, 193–202.
-
LeCun, Y., and Yoshua, B. (1995). Convolutional networks for images,speech, and time series. The Handbook of Brain Theory and Neural Net-works 3361, 255–258.
-
Bao, Y., Sivanandan, S., and Karaletsos, T. (2023). Channel Vision Trans-formers: An Image Is Worth c x 16 x 16 WordsThe Twelfth. InternationalConference on Learning Representations 4.
-
Kraus, O., Kenyon-Dean, K., Saberian, S., Fallah, M., McLean, P., Leung,J., Sharma, V., Khan, A., Balakrishnan, J., Celik, S., et al. (2024). Maskedautoencoders for microscopy are scalable learners of cellular biology. InIEEE Conference on Computer Vision and Pattern Recognition (CVPR),pp. 11757–11768.
-
Bao, Y., and Karaletsos, T. (2023). Contextual vision transformers forrobust representation learning. Preprint at arXiv.
-
Lopez, R., Regier, J., Cole, M.B., Jordan, M.I., and Yosef, N. (2018). Deepgenerative modeling for single-cell transcriptomics. Nature Methods 15,1053–1058.
-
Theodoris, C.V., Xiao, L., Chopra, A., Chaffin, M.D., Al Sayed, Z.R., Hill,M.C., Mantineo, H., Brydon, E.M., Zeng, Z., Liu, X.S., et al. (2023). Trans-fer learning enables predictions in network biology. Nature 618, 616–624.
-
Kobayashi-Kirschvink, K.J., Comiter, C.S., Gaddam, S., Joren, T., Grody,E.I., Ounadjela, J.R., Zhang, K., Ge, B., Kang, J.W., Xavier, R.J., et al.(2024). Prediction of single-cell RNA expression profiles in live cells byRaman microscopy with Raman2RNA. Nat. Biotechnol. 42, 1726–1734.
-
Ryu, J., Lopez, R., Bunne, C., and Regev, A. (2024). Cross-modalitymatching and prediction of perturbation responses with labeledGromov-Wasserstein optimal transport. Preprint at arXiv.
-
Saar, K.L., Scrutton, R.M., Bloznelyte, K., Morgunov, A.S., Good, L.L.,Lee, A.A., Teichmann, S.A., and Knowles, T.P.J. (2024). Protein Conden-sate Atlas from predic- tive models of heteromolecular condensatecomposition. Nat. Commun. 15, 5418.
-
Macosko, E.Z., Basu, A., Satija, R., Nemesh, J., Shekhar, K., Goldman,M., Tirosh, I., Bialas, A., Kamitaki, N., Martersteck, E., et al. (2015). Highlyparallel genome-wide expression profiling of individual cells using nano-liter droplets. Cell 161, 1202–1214.
-
Sta˚ hl, P.L., Salme´ n, F., Vickovic, S., Lundmark, A., Navarro, J.F., Mag-nusson, J., Giacomello, S., Asp, M., Westholm, J.O., Huss, M., et al.(2016). Visualization and analysis of gene expression in tissue sectionsby spatial transcriptomics. Science 353, 78–82.
-
Lundberg, E., and Borner, G.H.H. (2019). Spatial proteomics: a powerfuldiscovery tool for cell biology. Nat. Rev. Mol. Cell Biol. 20, 285–302.
-
Marconato, L., Palla, G., Yamauchi, K.A., Virshup, I., Heidari, E., Treis, T., Vierdag, W.M., Toth, M., Stockhaus, S., Shrestha, R.B., et al. (2024). Spa- tialData: an open and universal data framework for spatial omics. Nat. Methods. https://doi.org/10.1038/s41592-024-02212-x.
-
Somnath, V.R., Pariset, M., Hsieh, Y.-P., Martinez, M.R., Krause, A., andBunne, C. (2023). Aligned Diffusion Schro¨ dinger Bridges. In Uncertaintyin Artificial Intelligence, pp. 1985–1995.
-
Katharopoulos, A., Vyas, A., Pappas, N., and Fleuret, F. (2020). Fast au-toregressive transformers with linear attention. In International Confer-ence on Machine Learning.
-
Abramson, J., Adler, J., Dunger, J., Evans, R., Green, T., Pritzel, A., Ron-neberger, O., Willmore, L., Ballard, A.J., Bambrick, J., et al. (2024). Accu-rate structure prediction of biomolecular interactions with AlphaFold 3.Nature 630, 493–500.
-
Norman, T.M., Horlbeck, M.A., Replogle, J.M., Ge, A.Y., Xu, A., Jost, M.,Gilbert, L.A., and Weissman, J.S. (2019). Exploring genetic interactionmanifolds constructed from rich single-cell phenotypes. Science 365,786–793.
-
Lawson, M.J., Camsund, D., Larsson, J., Baltekin, O¨ ., Fange, D., and Elf,J. (2017). In situ genotyping of a pooled strain library after characterizingcomplex phenotypes. Mol. Syst. Biol. 13, 947.
-
Papamarkou, T., Skoularidou, M., Palla, K., Aitchison, L., Arbel, J., Dun-son, D., Filliponne, M., Fortuin, V., Hennig, P., Hernandez-Lobato, J.M.,et al. (2024). Position: bayesian deep learning is needed in the age oflarge-scale AI. In Forty-First International Conference on MachineLearning.
-
D’Angelo, F., and Fortuin, V. (2021). Wenzel F.On Stein Variational NeuralNetwork. Ensembles Preprint at arXiv.
-
Ober, S.W., Rasmussen, C.E., and van der Wilk, M. (2021). The promisesand pitfalls of deep kernel learning. In Conference on Uncertainty in Arti-ficial Intelligence, pp. 1206–1216.
-
Karaletsos, T. (2020). Bui T.D.Hierarchical Gaussian Process Priors forBayesian Neural Network Weights. Adv. Neural Inf. Process. Syst. 33,17141–17152.
-
Kapoor, S., Maddox, W.J., Izmailov, P., and Wilson, A.G. (2022). On un-certainty, tempering, and data augmentation in bayesian classification.Adv. Neural Inf. Process. Syst. 35, 18211–18225.
-
Lakshminarayanan, B., Pritzel, A., and Blundell, C. (2017). Simple andScalable Predictive Uncertainty Estimation using Deep Ensembles.Adv. Neural Inf. Process. Syst. 30.
-
Angelopoulos, A.N., and Bates, S. (2021). A gentle introduction toconformal prediction and distribution-free uncertainty quantification.Preprint at arXiv.
-
Cherian, J.J., Gibbs, I., and Cande` s, E.J. (2024). Large language modelvalidity via enhanced conformal prediction methods. Preprint at arXiv.
-
Cho, N.H., Cheveralls, K.C., Brunner, A.D., Kim, K., Michaelis, A.C., Ra-ghavan, P., Kobayashi, H., Savy, L., Li, J.Y., Canaj, H., et al. (2022). Open-Cell: endogenous tagging for the cartography of human cellular organiza-tion. Science 375, eabi6983.
-
Uhle´ n, M., Fagerberg, L., Hallstro¨ m, B.M., Lindskog, C., Oksvold, P.,Mardinoglu, A., Sivertsson, A˚ ., Kampf, C., Sjo¨ stedt, E., Asplund, A.,et al.. (2015). Proteomics. Tissue-based map of the human proteome.Science 347, 1260419.
-
Berger, C., Premaraj, N., Ravelli, R.B.G., Knoops, K., Lo´ pez-Iglesias, C.,and Peters, P.J. (2023). Cryo-electron tomography on focused ion beamlamellae transforms structural cell biology. Nat. Methods 20, 499–511.
-
Loconte, V., Chen, J.H., Vanslembrouck, B., Ekman, A.A., McDermott,G., Le Gros, M.A., and Larabell, C.A. (2023). Soft X-ray tomograms pro-vide a structural basis for whole-cell modeling. FASEB J. 37, e22681.
-
Moffitt, J.R., Lundberg, E., and Heyn, H. (2022). The emerging landscapeof spatial profiling technologies. Nat. Rev. Genet. 23, 741–759.
-
Vandereyken, K., Sifrim, A., Thienpont, B., and Voet, T. (2023). Methodsand applications for single-cell and spatial multi-omics. Nat. Rev. Genet.24, 494–515.
-
Tabula Sapiens Consortium*, Jones, R.C., Karkanias, J., Krasnow, M.A.,Pisco, A.O., Quake, S.R., Salzman, J., Yosef, N., Bulthaup, B., Brown, P.,et al. (2022). The tabula sapiens: A multiple-organ, single-cell transcrip-tomic atlas of humans. Science 376, eabl4896.
-
He, B., Bergenstra˚ hle, L., Stenbeck, L., Abid, A., Andersson, A., Borg, A˚ .,Maaskola, J., Lundeberg, J., and Zou, J. (2020). Integrating spatial geneexpression and breast tumour morphology via deep learning. Nat. Bio-med. Eng. 4, 827–834.
-
Bock, C., Boutros, M., Camp, J.G., Clarke, L., Clevers, H., Knoblich, J.A.,Liberali, P., Regev, A., Rios, A.C., Stegle, O., et al. (2021). The organoidcell atlas. Nat. Biotechnol. 39, 13–17.
-
Tabula; Muris Consortium; Overall coordination; Logistical coordination;Organ collection and processing; Library preparation and sequencing;Computational data analysis; Cell type annotation; Writing group; Sup-plemental text writing group; Principal investigators (2018). Single-celltranscriptomics of 20 mouse organs creates a tabula muris. Nature562, 367–372.
-
Li, H., Janssens, J., De Waegeneer, M., Kolluru, S.S., Davie, K., Gardeux,V., Saelens, W., David, F.P.A., Brbic´, M., Spanier, K., et al. (2022). Fly CellAtlas: A single-nucleus transcriptomic atlas of the adult fruit fly. Science375, eabk2432.
-
Lange, M., Granados, A., Vijaykumar, S., Bragantini, J., Ancheta, S., San-thosh, S., Borja, M., Kobayashi, H., McGeever, E., Solak, A.C., et al.(2023). Zebrahub – Multimodal zebrafish Developmental Atlas Revealsthe State Transition Dynamics of Late Vertebrate Pluripotent Axial Pro-genitors. Preprint at bioRxiv.
-
Katz, K., Shutov, O., Lapoint, R., Kimelman, M., Brister, J.R., and O’Sul-livan, C. (2022). The Sequence Read Archive: a decade more of explosivegrowth. Nucleic Acids Res. 50, D387–D390.
-
Achiam, J., et al. (2023). GPT-4 technical report. Preprint at arXiv.
-
Ding, F., and Steinhardt, J.N. (2024). Protein language models are biasedby unequal sequence sampling across the tree of life. Preprint at bioRxiv.
-
Liao, W.-W., Asri, M., Ebler, J., Doerr, D., Haukness, M., Hickey, G., Lu,S., Lucas, J.K., Monlong, J., Abel, H.J., et al. (2023). A draft human pan-genome reference. Nature 617, 312–324.
-
Liu, J., Shen, Z., He, Y., Zhang, X., Xu, R., Yu, H., and Cui, P. (2021). To-wards out-of-distribution generalization: A survey. Preprint at arXiv.
-
Nisonoff, H., Wang, Y., and Listgarten, J. (2023). Coherent blend- ing of biophysics-based knowledge with bayesian neural networks for robust protein property prediction. ACS Synth. Biol. 12, 3242–3251. https://doi.org/10.1021/acssynbio.3c00217.
-
Zheng, F., Kelly, M.R., Ramms, D.J., Heintschel, M.L., Tao, K., Tutuncuo-glu, B., Lee, J.J., Ono, K., Foussard, H., Chen, M., et al. (2021). Interpre-tation of cancer mutations using a multiscale map of protein systems.Science 374, eabf3067.
-
Ma, J., Yu, M.K., Fong, S., Ono, K., Sage, E., Demchak, B., Sharan, R.,and Ideker, T. (2018). Using deep learning to model the hierarchical struc-ture and function of a cell. Nat. Methods 15, 290–298.
-
Gao, S., Fang, A., Huang, Y., Giunchiglia, V., Noori, A., Schwarz, J.R., Ek-tefaie, Y., Kondic, J., and Zitnik, M. (2024). Empowering biomedical dis-covery with AI agents. Cell 187, 6125–6151.
-
Hurrell, T., Naidoo, J., Ntlhafu, T., and Scholefield, J. (2024). An Africanperspective on genetically diverse human induced pluripotent stem celllines. Nat. Commun. 15, 8581.
-
Gibbs, R.A., Belmont, J.W., Hardenbol, P., Willis, T.D., Yu, F.L., Yang,H.M., Ch’ang, L.Y., Huang, W., Shen, B., Tam, Y., et al. (2003). The inter-national HapMap project. Nature 5, 467–475.
-
Cancer; Genome; Atlas; Research Network, Weinstein, J.N., Collisson,E.A., Mills, G.B., Shaw, K.R.M., Ozenberger, B.A., Ellrott, K., Shmulevich,I., Sander, C., and Stuart, J.M. (2013). The cancer genome atlas pan-can-cer analysis project. Nat. Genet. 45, 1113–1120.
-
ENCODE Project Consortium (2012). An integrated encyclopedia of DNAelements in the human genome. Nature 489, 57–74.
-
Lonsdale, J., Thomas, J., Salvatore, M., Phillips, R., Lo, E., Shad, S.,Hasz, R., Walters, G., Garcia, F., Young, N., and Foster, B. (2013). TheGenotype-Tissue Expression (GTEx) project. Nat. Genet. 45, 580–585.
-
Ponte´ n, F., Jirstro¨ m, K., and Uhlen, M. (2008). The Human Protein Atlas–atool for pathology. J. Pathol. 216, 387–393.
-
Downey, P., and Peakman, T.C. (2008). Design and implementation of ahigh-throughput biological sample processing facil- ity using modernmanufacturing principles. Int. J. Epidemiol. 37 (Suppl 1 ), i46–i50.
-
Madani, A., Krause, B., Greene, E.R., Subramanian, S., Mohr, B.P., Hol-ton, J.M., Olmos, J.L., Xiong, C., Sun, Z.Z., Socher, R., et al. (2023). Largelanguage models generate func- tional protein sequences across diversefamilies. Nat. Biotechnol. 41, 1099–1106.
-
Nelson, M.R., Tipney, H., Painter, J.L., Shen, J., Nicoletti, P., Shen, Y.,Floratos, A., Sham, P.C., Li, M.J., Wang, J., et al. (2015). The supportof human genetic evidence for approved drug indications. Nat. Genet.47, 856–860.
-
Mason, C., Brindley, D.A., Culme-Seymour, E.J., and Davie, N.L. (2011).Cell therapy industry: billion dollar global business with unlimited poten-tial. Regen. Med. 6, 265–272.
-
Bashor, C.J., Hilton, I.B., Bandukwala, H., Smith, D.M., and Veiseh, O.(2022). Engineering the next generation of cell-based therapeutics. Nat.Rev. Drug Discov. 21, 655–675.
-
Jia, Q., Wang, A., Yuan, Y., Zhu, B., and Long, H. (2022). Heterogeneity ofthe tumor immune microenvironment and its clinical relevance. Exp.Hematol. Oncol. 11, 24.
-
Melssen, M.M., Sheybani, N.D., Leick, K.M., and Slingluff, C.L. (2023).Barriers to immune cell infiltration in tumors. J. Immunother. Cancer 11.
-
Chow, A., Perica, K., Klebanoff, C.A., and Wolchok, J.D. (2022). Clinicalimplications of T cell exhaustion for cancer immunotherapy. Nature Re-views Clinical Oncology 19, 775–790.
-
de Visser, K.E., and Joyce, J.A. (2023). The evolving tumor microenviron-ment: from cancer initiation to metastatic outgrowth. Cancer Cell 41,374–403.
-
Barkley, D., Moncada, R., Pour, M., Liberman, D.A., Dryg, I., Werba, G.,Wang, W., Baron, M., Rao, A., Xia, B., et al. (2022). Cancer cell statesrecur across tumor types and form specific interactions with the tumormicroenvironment. Nat. Genet. 54, 1192–1201.
-
Schwartzberg, L., Kim, E.S., Liu, D., and Schrag, D. (2017). Precisiononcology: who, how, what, when, and when not? American Society ofClinical Oncology Educational Book 37, 160–169.
-
Aebersold, R., Agar, J.N., Amster, I.J., Baker, M.S., Bertozzi, C.R., Boja,
E.S., Costello, C.E., Cravatt, B.F., Fenselau, C., Garcia, B.A., et al. (2018).How many human proteoforms are there? Nat. Chem. Biol. 14, 206–214.
-
Katsoulakis, E., Wang, Q., Wu, H., Shahriyari, L., Fletcher, R., Liu, J.,Achenie, L., Liu, H., Jackson, P., Xiao, Y., et al. (2024). Digital twins forhealth: a scoping review. npj Digit. Med. 7, 77.
-
Rajewsky, N., Almouzni, G., Gorski, S.A., Aerts, S., Amit, I., Bertero,M.G., Bock, C., Bredenoord, A.L., Cavalli, G., Chiocca, S., et al. (2020).Lifetime and improving European healthcare through cell-based inter-ceptive medicine. Nature 587, 377–386.
-
Alix-Panabie` res, C., and Pantel, K. (2021). Liquid biopsy: from discoveryto clinical application. Cancer Discov. 11, 858–873.
-
Vaishnav, E.D., de Boer, C.G., Molinet, J., Yassour, M., Fan, L., Adiconis,X., Thompson, D.A., Levin, J.Z., Cubillos, F.A., and Regev, A. (2022). Theevolution, evolvability and engineering of gene regulatory DNA. Nature603, 455–463.
-
Go´ mez-de-Mariscal, E., Garcı´a-Lo´ pez-de-Haro, C., Ouyang, W., Donati,L., Lundberg, E., Unser, M., Mun˜ oz-Barrutia, A., and Sage, D. (2021).DeepImageJ: A user-friendly environment to run deep learning modelsin ImageJ. Nat. Methods 18, 1192–1195.
-
Le, T., Winsnes, C.F., Axelsson, U., Xu, H., Mohanakrishnan Kaimal, J.,Mahdessian, D., Dai, S., Makarov, I.S., Ostankovich, V., Xu, Y., et al.(2022). Analysis of the human protein atlas weakly supervised single-cell classification competition. Nat. Methods 19, 1221–1229.
-
Chen, R.J., Ding, T., Lu, M.Y., Williamson, D.F.K., Jaume, G., Song, A.H.,Chen, B., Zhang, A., Shao, D., Shaban, M., et al. (2024). Towards a gen-eral-purpose foundation model for computational pathology. Nat. Med.30, 850–862.
-
Moen, E., Bannon, D., Kudo, T., Graf, W., Covert, M., and Van Valen, D.(2019). Deep learning for cellular image analysis. Nat. Methods 16,1233–1246.
-
Avsec, Zˇ., Weilert, M., Shrikumar, A., Krueger, S., Alexandari, A., Dalal,K., Fropf, R., McAnany, C., Gagneur, J., Kundaje, A., et al. (2021).Base-resolution models of transcription-factor binding reveal soft motifsyntax. Nat. Genet. 53, 354–366.
-
Ho, J., Jain, A., and Abbeel, P. (2020). Denoising diffusion proba- bilisticmodels. Adv. Neural Inf. Process. Syst. 33, 6840–6851.
-
Lipman, Y., Chen, R.T., Ben-Hamu, H., Nickel, M., and Le, M. (2023).Flow Matching for Generative Modeling. International Conference onLearning Representations.
-
Scarselli, F., Gori, M., Tsoi, A.C., Hagenbuchner, M., and Monfardini, G.(2009). The graph neural network model. IEEE Trans. Neural Netw. 20,61–80.
-
Cao, Y., and Shen, Y. (2020). Energy-based graph convolutional net-works for scoring protein docking models. Proteins 88, 1091–1099.
-
Brbic´, M., Cao, K., Hickey, J.W., Tan, Y., Snyder, M.P., Nolan, G.P., andLeskovec, J. (2022). Annotation of spatially resolved single-cell data withSTELLAR. Nat. Methods 19, 1411–1418.
-
Wu, Z., Trevino, A.E., Wu, E., Swanson, K., Kim, H.J., D’Angio, H.B., Pre-ska, R., Charville, G.W., Dalerba, P.D., Egloff, A.M., et al. (2022). Graphdeep learning for the characterization of tumour microenvironmentsfrom spatial protein profiles in tissue specimens. Nat. Biomed. Eng. 6,1435–1448.
-
Hamilton, W., Ying, Z., and Leskovec, J. (2017). Inductive RepresentationLearning on Large Graphs. Adv. Neural Inf. Process. Syst. 30.
参考文献可上下滑动查看
生命复杂性读书会:
生命复杂系统的构成原理
在生物学中心法则的起点,基因作为生命复杂系统的遗传信息载体,在生命周期内稳定存在;而位于中心法则末端的蛋白质,其组织构成和时空变化的复杂性呈指数式增长。随着分子生物学数十年来的突飞猛进,尤其是生命组学(基因组学、转录组学、蛋白质组学和代谢组学等的集合)等领域的日新月异,当代生命科学临近爆发的边缘。如此海量的数据如何帮助我们揭示宇宙中最复杂的物质系统——“人体”的构成原理和设计原理?阐释人类发育、衰老和重大疾病的发生机制?
集智俱乐部联合西湖大学理学院及交叉科学中心讲席教授汤雷翰,国家蛋白质科学中心(北京)副研究员常乘、李杨,香港浸会大学助理教授唐乾元,北京大学前沿交叉学科研究院研究员林一瀚,中国科学院分子细胞科学卓越创新中心博士后唐诗婕,共同发起「生命复杂性:生命复杂系统的构成原理」读书会,从微观细胞尺度、介观组织器官尺度到宏观人体尺度,梳理生命科学领域中的重要问题及重要数据,由生物学家提问,希望促进统计物理、机器学习方法研究者和生命科学研究者之间的深度交流,建立跨学科合作关系,激发新的研究思路和合作项目。读书会目前共进行10期,现在报名参与读书会可以加入读书会社群,观看视频回放,解锁完整读书会权限。
7. 重整化群与非线性物理,寻找复杂系统跨尺度的分析方法丨新课发布
微信扫一扫,分享到朋友圈











