Physics Reports计算网络生物学长文综述:数据、模型和应用

生物网络是网络科学的重要研究方向,蛋白质网络等研究已经在新冠肺炎药物搜索中发挥了作用。近日由阿里巴巴复杂科学研究中心张子柯、美国克利夫兰医学中心程飞雄、瑞士弗里堡大学张翼成等人组成的研究团队,在物理学顶刊 Physics Reports 发表综述文章,全面介绍了常见生物网络类型、网络结构、分析方法以及生物医学应用等方面的相关研究。本文将深入解读这篇综述的主要内容。
我们特邀论文第一作者、杭州师范大学阿里巴巴复杂科学研究中心刘闯老师,在北京时间 5 月 27 日(周三)19:00-20:00 做客集智俱乐部直播间,亲述论文故事,报名方式见文末。

论文题目: Computational network biology: Data, models, and applications
论文地址: https://www.sciencedirect.com/science/article/abs/pii/S0370157319304041
生物网络是网络科学研究的重要方向,其相关研究伴随着网络科学的发展和生物数据技术的进步,在近年来取得了突飞猛进的进展。生物网络是以生物分子作为节点,分子之间的相互关联为连边构成的系统(如图1所示)。生物网络种类繁多,比较常见的主要有蛋白-蛋白互作网络、基因调控网络、基因关联网络、代谢网络、脑网络等等。本文将以疾病相关的生物网络计算研究为主要对象,从下述几个方面进行介绍。
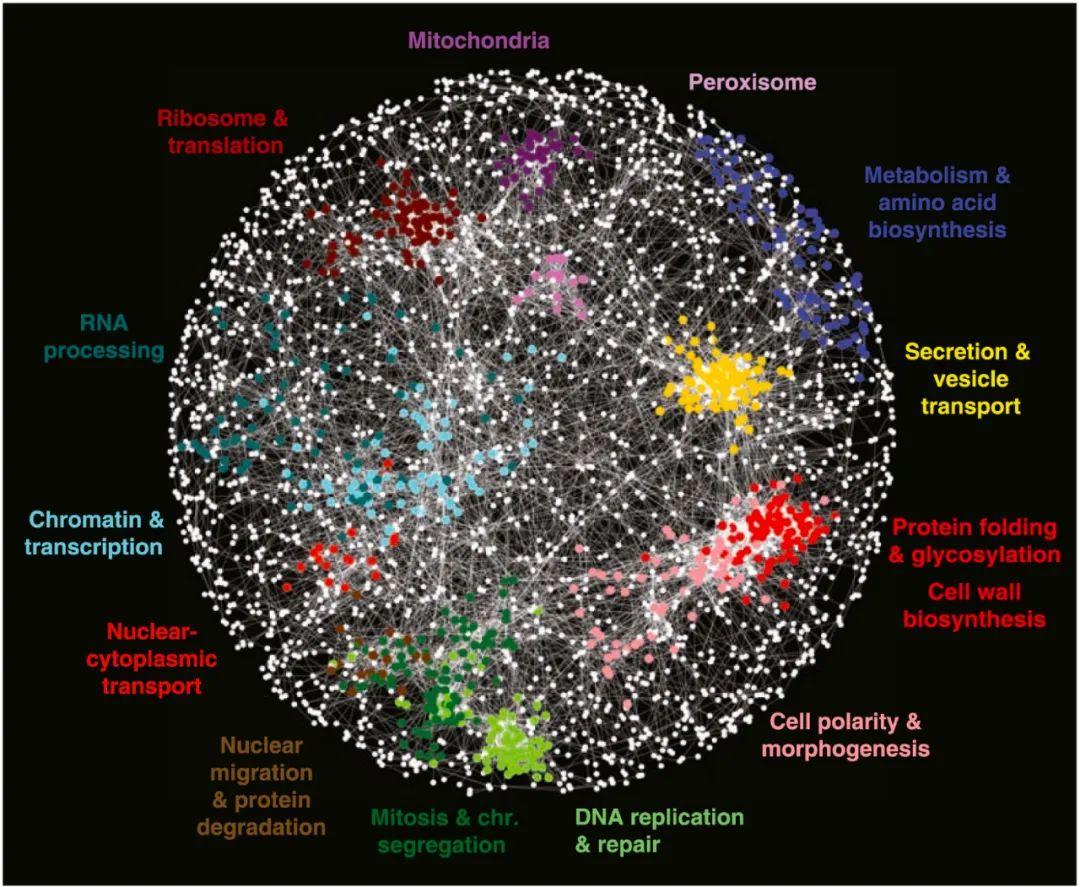
1. 网络结构与生物功能
生物网络结构复杂,对生物网络结构的分析,有助于我们更好的理解生物功能。生物网络具有很强的异质性,网络度分布大多呈现幂律分布特征。Jeong 等[2]在蛋白-蛋白网络中发现的中心性-致死率准则(Centrality-Lethality Rule),即蛋白质的重要性与其在蛋白-蛋白网络中的中心性高度相关,类似的发现也在多种生物分子网络中被证实。
大量研究表明生物分子并不是独自完成其生物功能,而是通过与其他生物分子之间的相互作用,形成社团结构来表达其功能。例如,在蛋白-蛋白网络中,具有特定功能的蛋白、形成蛋白复合物的蛋白、同一种疾病的致病蛋白等都倾向于形成社团(如图1所示);在代谢网络中,处于同一代谢通路上的代谢物更倾向于形成社团;在脑功能网络中,具有特定认知功能的神经元也倾向于形成社团。特别地,生物网络的社团特征还会表现出比较强的层次结构[3]。
正常组织和疾病状态下的网络结构一般会呈现出明显差异,对于这种结构特征差异的捕捉有助于理解疾病的发生及演化。例如,在脑功能网络中,老年痴呆患者的脑功能网络的网络效率显著降低,并且网络的小世界特性也随之消失;当大脑从脑外伤中恢复的过程中,脑网络逐步朝着网络效率更高的方向演化。肿瘤细胞网络的结构熵明显高于正常网络,并且随着不同阶段的演化,不同肿瘤类型呈现出类似的结构熵演化模式。然而,目前对于生物网络结构随着生物进化的演化过程以及生物网络如何影响细胞功能等核心问题尚没有完善的研究成果。
2. 关键生物分子识别
在网络分析中,网络中心性是刻画节点重要性的关键指标,该假设在生物网络中同样适用。网络中心性较高的生物分子通常都与疾病发生、药物靶点等相关 [4]。在生物网络中,通常单个基因突变可以通过网络传播来放大突变信号,进而扰动整个通路导致疾病发生[5]。因此,网络传播,尤其是带重启的随机游走方法(Random Walk with Restart),被广泛应用于关键生物分子的预测。例如,在疾病基因预测中,给已知疾病基因赋予一定的分数,然后采用网络传播的方法,在系统达到稳态时,分数较高的基因也是相应的疾病基因的概率较高 [6]。Leiserson 等提出的 HotNet 方法利用肿瘤病人的基因突变信号在网络中的传播来识别肿瘤的驱动基因,算法发现了一些罕见突变的肿瘤驱动基因[7]。此外,网络传播方法也可以用于弥补生物临床数据的稀疏性,为深入分析提供更平滑的数据[8]。
网络结构可控性分析也有助于识别关键生物分子。Vinayagam 等基于网络可控性得到了蛋白-蛋白网络的不可或缺节点集(Indispensable),即删除这些节点会导致控制节点个数增加,这些不可或缺节点都是常见的致病突变或者人类病毒的目标蛋白和一些已知的药物靶标[9]。Wuchty 通过最小支配点集的方法找到网络的控制蛋白,发现这些蛋白不仅具有重要的功能(如癌症相关蛋白、病毒目标蛋白等),而且在网络信号控制中也扮演重要角色(如转录因子、激酶蛋白)[10]。此外,基于生物实验的研究结果也进一步验证了网络控制在关键生物分子识别中的作用 [11]。
3. 网络疾病
疾病通常很少是由于单个基因异常,而是组织和器官系统的复杂生物网络的扰动或故障[12]而引起的。网络医学的假设认为,基因变异(包括扩增、缺失、移位和突变等)导致生物分子网络发生扰动,进而使得网络性质和系统状态发生改变,而导致疾病的发生发展维持和恶化[12]。这一假设也被功能学研究所证实,疾病相关的等位基因变异通常改变的是蛋白-蛋白相互作用,而不是蛋白质的折叠和稳定性[13]。
因此,大多数疾病更应该被描述为“网络疾病”。在这个层面上说,基于边突变(Edgetic mutation)[13]和网络突变(Network-attacking mutation)[14]的描述范式,更有助于从人类蛋白-蛋白相互作用的生物分子水平更好地评估人类疾病的内在复杂性。
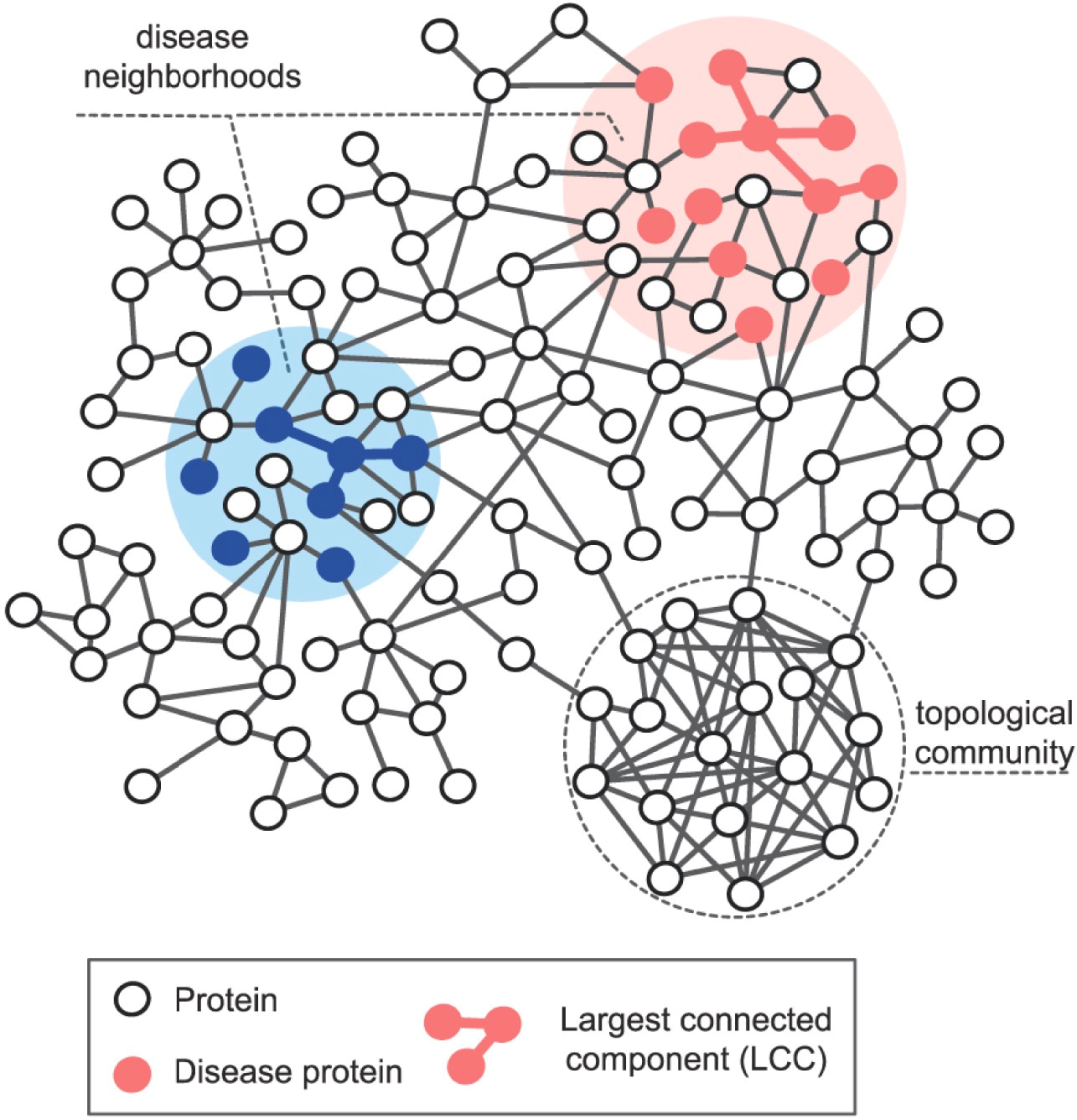
对于疾病蛋白的研究表明,疾病蛋白之间更倾向于形成蛋白-蛋白相互作用,编辑这组蛋白的基因也更倾向于在特定的组织中共表达[15]。因此,疾病蛋白并不是随机的分布在蛋白-蛋白网络中,而是会相互之间连接形成一个或几个子网络,这被定义为疾病模块(如图2所示)。Menche 等研究了 299 种疾病,通过最大连通子集和疾病直径(每个疾病蛋白到与其距离最近的疾病蛋白之间的距离的平均值)的分析发现其中 226 种疾病蛋白显著倾向于形成疾病模块。
疾病模块的发现不仅有助于从系统性的视角理解疾病发生发展,也能够促进疾病药物的研发。基于网络的计算研究为疾病模块挖掘提供了大量的方法[16],按照数据场景大致可以分为两类:(1)基于已知的疾病基因,寻找网络中最有可能与其构成模块的基因集,进而形成疾病模块[17];(2)整合生物网络数据和疾病相关的基因组学数据,包括差异表达、突变序列等,定义相应的目标函数,采用网络搜索的方法寻找疾病模块[18]。
4. 基于药物-基因-疾病
网络的药物研发
虽然最近几十年生物医学和制药学的研究取得了长足的发展,但是每年新增的被 FDA 认证的药物治疗方案依然非常有限,其中一个主要原因就是在目前的药物研发中依然遵循“一个药物、一个基因、一种疾病”的模式。然而大多数复杂疾病都是多基因作用导致的,网络药理学认为单一靶标的干预远远不足以对复杂系统性疾病产生好的效果,因此药物研发应该从单一靶标向“网络靶向-多组分治疗”的模式转变[19],生物网络研究为基于多靶标的药物研发提供了基础。
药物靶标预测。药物靶标的作用关系是药物研发和设计的基础,但是药物靶标作用的实验是非常耗费资源的。因此,基于计算辅助的药物靶标预测方法可以发现药物的潜在靶点,为实验验证发现新的药物疗效提供理论基础。
常见的药物靶标预测是基于相似性的假设,比如对于一个已知的药物-靶标对,与该药物很相似的药物也有很大的概率与该靶标产生作用,与该靶标相似的靶标有很大的概率与该药物产生作用。通过构建药物-靶标二部分图,利用目标药物的已知靶标上设置的分数在网络中扩散,最后将靶标按所得分数进行排序,排在前面的靶标就可能是该药物的潜在靶标[20]。最近,Zeng 等整合多维数据,包括药物-靶标关系、蛋白-蛋白作用关系、药物-副作用关系、药物-药物相似度、靶标-靶标相似度等,开发了基于网络的深度学习方法进行药物靶标预测,显著提升了预测准确度[21]。
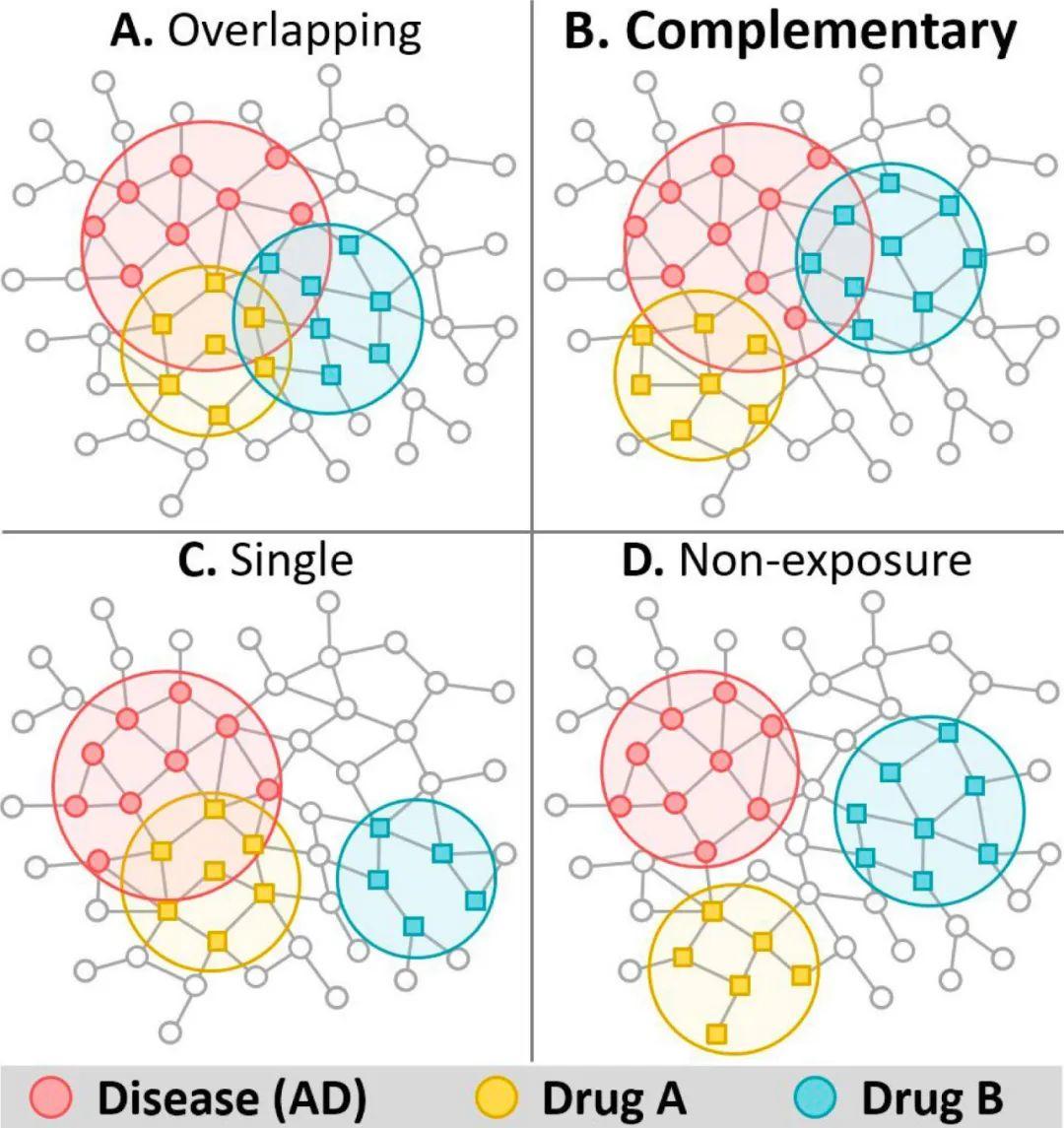
旧药新用。新药由于其研发成本高、时间长,因此效率相对较低,旧药新用是目前药物研发的一种有效方案。旧药新用是对于已经上市的药物,发现其新的疗效,而用于其他疾病的治疗。基于网络医学的旧药新用的两个基本假设是(1)疾病相关的蛋白(或药物的靶点蛋白)都倾向于形成一个相互连接的疾病(或药物靶向)模块和(2)药物靶向模块与疾病模块在网络上显著接近,则该药物对疾病可能有疗效(如图3所示)。基于药物靶向蛋白和疾病蛋白的网络距离而定义的网络亲密度(Network-based drug–disease proximity)是一种简单常用的刻画药物疾病关系的方法。研究发现最有效的网络距离的定义如下[22]:
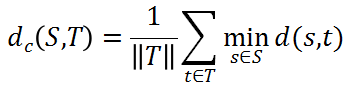
其中,t∈T 表示药物的靶点蛋白集合,s∈S 表示疾病的相关蛋白集合,d(s,t)表示药物靶点 t 与疾病蛋白 s 之间的最短路径长度,|| T || 表示药物的靶点蛋白的数目。
药物疾病的网络亲密度就是上述距离相较于两个同等规模和度序列的随机模块之间距离分布的显著性(用 Z-score 来表示),该距离显著低于随机模块之间的距离(例如 Z-score<-2),则该药物就为该疾病潜在的候选药物。Cheng等通过整合个体基因突变数据、基因共表达数据和人体蛋白-蛋白作用网络数据,提出了一种全基因组定位系统网络算法(Genome-wide Positioning Systems network)挖掘疾病蛋白模块,并通过计算该特定疾病模块与药物靶标之间的网络亲进度进行药物重定位研究,以非小细胞肺癌为例发现了一种新的潜在药物(乌苯苷)及其作用靶点[23]。
对于目前肆虐的新冠肺炎病毒,基于生物网络分析的旧药新用方法也为药物研发提供了支持,通过选定的受新冠病毒影响的人体蛋白质(疾病蛋白)与药物靶标之间的关系,筛选出一系列潜在的候选药物,为后续的生物试验与临床测试提供理论基础 [24,25]。
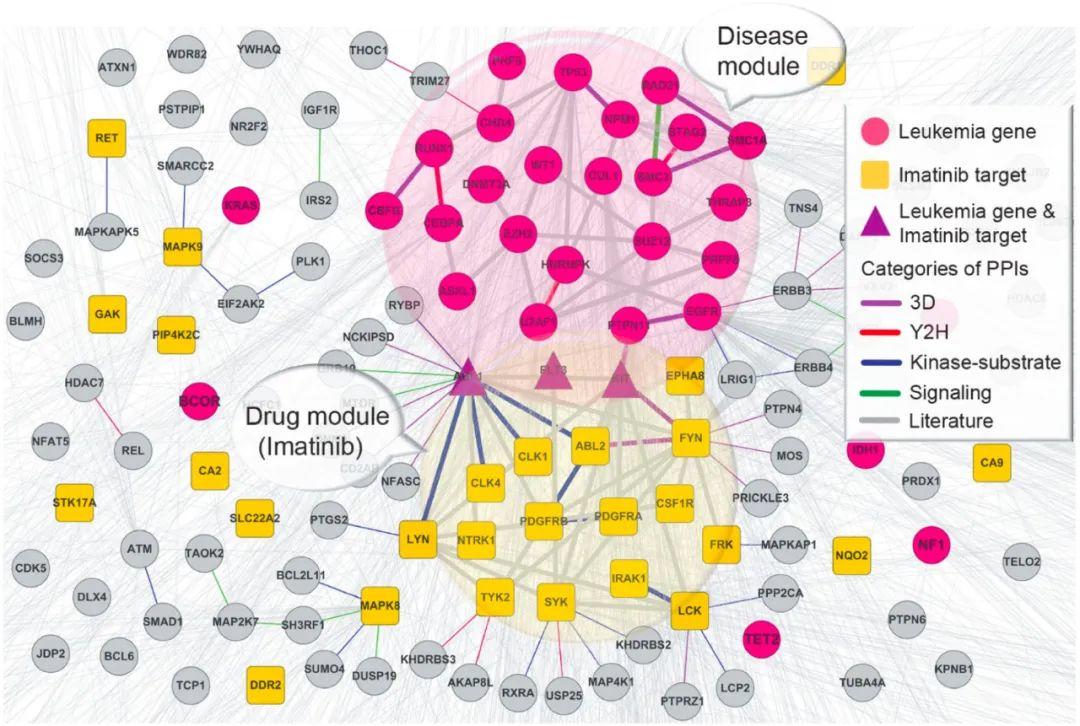
药物组合。通常与单一药物疗法相比,药物组合(联合疗法)通过使用两种或多种药物以协同靶向多种疾病蛋白或通路,显示出更高的疗效。同时,药物组合还由于每种活性成分的低剂量,可以减少毒副作用。因此,在疾病治疗中确定高效低毒的药物组合方案显得尤为重要。基于生物网络的药物组合研究表明,药物组合具有治疗增强效果必须满足两个原则:(1)两种药物靶向模块必须与疾病模块重叠,(2)两种药物靶向模块必须在蛋白-蛋白网络中相互分离,而不重叠毒性机制(如图4B所示)[26]。模块与之间的重叠和分离的可用下述公式进行度量:

其中,<dAB>,<dAA>,<dBB>和分别表示模块 AB 之间,模块 A 以及模块 B 之内的节点对的平均最短距离。S<0 表示两个模块是重叠关系,S>0 表示两个模块是分离关系。相关研究方法也应用于新冠肺炎病毒的药物组合治疗研究中[27]。
展望与挑战
近年来,计算生物网络在理论和方法都取得了丰硕的成果,并广泛应用于理解生物分子的内在联系、疾病的发现及治疗和药物的研发等等。论文主要梳理了如下几个方面挑战与发展方向。(1)目前的生物网络数据存在着不平衡不完整的缺陷,高质量的生物网络数据获取方法,也是生物网络研究的重要基础。(2)生物网络包含多种生物分子不同类型的连接,为异构网络以及网络的网络等方面的研究提供了良好的场景。(3)生物网络数据复杂度高,基于机器学习特别是深度学习的研究应用,将为多维度的生物网络数据分析带来新的机遇。(4)基于生物试验的假设与验证对于生物网络研究也尤为重要,这都需要不同学科的研究人员从不同角度为该领域的发展做出贡献。
(参考文献可上下滑动)
作者:刘闯
编辑:张爽
集智科学家直播预告
我们邀请到论文第一作者杭州师范大学刘闯老师,在北京时间5月27日(周三)19:00-20:00,做客集智俱乐部直播间,从研究者角度来解读这篇论文。


扫描二维码,点击“我要听”报名Zoom会议室直播
推荐阅读

集智俱乐部QQ群|877391004
商务合作及投稿转载|swarma@swarma.org
◆ ◆ ◆
搜索公众号:集智俱乐部
加入“没有围墙的研究所”

让苹果砸得更猛烈些吧!
👇点击文末“阅读原文”报名「我要听」,提前向讲者提问
微信扫一扫,分享到朋友圈











