系统生物学及相关跨学科领域正在兴起,近年来基于各类疾病的组学研究成果频出。作者整理了这份结合多门教材、多篇经典论文的学习路径,供你入门参考。如果你希望入门系统生物学,请扫开头二维码或点击文末“阅读原文”,注册集智斑图,资料更完备的学习路径等着你~
https://pattern.swarma.org/path?id=67&from=wechat
系统生物学(Systems biology),是一个使用整体论(而非还原论)研究范式,整合不同学科、层次的信息以理解生物系统如何行使功能的学术领域,是分子生物学之后现代生物学的全新阶段,包括表观遗传学、各种生物组学、合成生物学、生物信息学等细分领域。
它通过融合数学、物理、化学、生物、医学、信息与计算科学等多学科方法,从一种全新的生物动力学视角出发对生命现象进行研究,包括分子、细胞、器官、生物有机体乃至环境等实体生物系统各个组成部分相互作用关系下的表型、功能和行为。因此它兼具生物学和信息科学的特点。
一般说来,生物信息以这样的方向进行流动:DNA→mRNA→蛋白质→蛋白质相互作用网络→细胞→器官→个体→群体。这里要注意的是,每个层次信息都对理解生命系统的运行提供有用的视角。不同层次的研究难度也是不一样的。系统生物学的重要任务就是要尽可能地获得每个层次的信息并将它们进行整合。
生物有机体是非常复杂的,许多部分以多种方式相互作用,因此通常可以被看作是一个集成的系统。从这点看一个细胞信号网络和一个收音机有很多相似之处[1]。但比之收音机,试图理解一个生物有机体系统要困难得多,主要是因为系统中交互作用的数量和强度过于巨大且缺乏一种通用的生物学描述语言。不过,可用的计算机能力和复杂系统分析的进步带来了希望,成为系统生物学的基本和不可缺少的方法。
Can a biologist fix a radio?—Or, what I learned while studying apoptosis
https://doi.org/10.1007/s10541-005-0088-1
系统生物学不同于以往仅仅关心个别的基因和蛋白质的分子生物学,着眼于研究细胞信号传导和基因调控网路、生物系统组成之间相互关系的结构和系统功能的涌现。系统生物学的目标之一就是模拟和发现涌现的特性,并期望最终能够建立包括整个生物系统的可理解模型。
事实上,据 2011 年的《cell》报导,这一壮举已经在生殖支原体(Mycoplasma genitalium)的细胞模型中实现了,其中所有的基因、产物以及已知的代谢相互作用都已在电脑中重建[2]。
A whole-cell computational model predicts phenotype from genotype
https://www.sciencedirect.com/science/article/pii/S0092867412007763
也许我们很快就会看到一个完整的多细胞生物的电脑模拟模型。尽管这对于数百万到数万亿个细胞来说似乎是不可行的,但我们仍对科学的发展满怀热情,毕竟目前所取得的研究成果,10 年前在计算或技术上也曾被认为是不可能的。
1924-1928 年奥地利生物学家贝塔郎菲多次发表系统论的文章,阐述生物学中有机体概念,提出把有机体当作一个整体或系统来研究。贝塔朗菲的一般系统论,维纳的控制论到香农的信息论,及普利高津的耗散结构理论(Dissipative structures),均将生命现象看作区别于仅靠外部指令运作的自组织系统。
自 20 世纪 60 年代系统生物学概念和词汇的提出起,60-80 年代系统生态学、系统生理学的进展,90 年代系统生物医学、系统医学、系统生物工程与系统遗传学的概念发表,与 20 世纪未细胞信号传导与基因调控的研究与系统论方法的结合后,系统生物学进入了分子细胞层次的(实验与理论结合)研究与发展时期。尤其在 1979 年,英国生物化学家艾根提出超循环理论(Hypercycles),对无机分子自组织成生物大分子的可能机制进行了解释,打通了生命系统和无机物之间的桥梁。
21世纪,系统生物学的发展进入了细胞信号转导与基因表达调控的细胞分子系统生物学时期,国际,国内的系统与合成生物学,系统遗传学等研究机构纷纷建立,让生物学进入了系统生命科学时代。2001 年的第二届国际系统生物学会提出对生物体整个过程做全面性的定量研究,并希望利用计算机运算来预测细胞,器官系统甚至完整生物体的表现。
人类基因组计划(HGP:Human Genome Project ; 1990–2003)的发起人之一,美国科学家莱诺伊·胡德 (Leroy Hood) 是组学 (Omics) 生物技术开创者之一。正是在基因组学(Genomics)、蛋白质组学(Proteomics)等新型大科学发展的基础上,孕育了系统生物学种种高通量生物技术和生物信息技术。
1. 初步模型:
是对选定的某一生物系统的所有组分进行了解和确定,描绘出该系统的结构,包括基因相互作用网络和代谢途径,以及细胞内和细胞间的作用机理,以此构造出一个初步的系统模型。
2. 观测实验:
是系统地改变被研究对象的内部组成成分(如基因突变)或外部生长条件,然后观测在这些情况下系统组分或结构所发生的相应变化,包括基因表达、蛋白质表达和相互作用、代谢途径等的变化,并把得到的有关信息进行整合。
3. 分析修订:
把通过实验得到的数据与根据模型预测的情况进行比较,并对初始模型进行修订。
4. 理想模型:
根据修正后的模型的预测或假设,设定和实施新的改变系统状态的实验,重复第二步和第三步,不断地通过实验数据对模型进行修订和精练。第一到第三阶段,也就是所谓的“整合”- 系统理论、“干涉”- 实验生物学和“信息”- 计算生物学研究过程,即系统生物学通过系统论和实验(Experimental)、计算(Computational)等概念和方法的整合,目标就是要得到一个理想的完整模型,使其理论预测能够全面反映出生物系统的真实性。
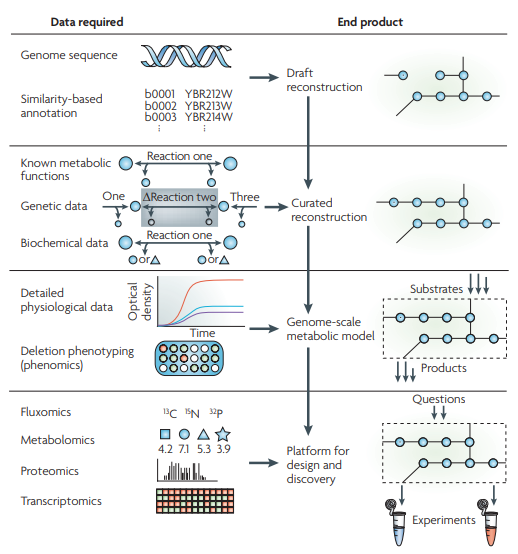
以上图四个阶段为例,每一个阶段都是在前一个阶段的基础上建立起来的。重构过程的另一个特点是重构内容的迭代细化,这是由后三个阶段的实验数据驱动的。对于每个阶段,都需要特定的数据类型,这些数据类型包括从高通量数据类型(例如,基因组学和代谢组学)到描述单个成分的详细研究(例如,特定反应的生化数据)。每个重构阶段生成的结果可以用于检查越来越多的问题,最终获得理想模型[3]。
Reconstruction of Biochemical Networks in Microorganisms
https://www.nature.com/articles/nrmicro1949
凡是实验科学都有这样一种特征:人为地设定某种或某些条件去作用于被实验的对象,从而达到实验的目的。这种对实验对象的人为影响就是干涉 (Perturbation)。系统生物学中的干涉有这样一些特点。首先,这些干涉应该是有系统性的。例如人为诱导基因突变,过去大多是随机的;而在进行系统生物学研究时,应该采用的是定向的突变技术。
以测定基因组全序列或全部蛋白质组成的基因组研究或蛋白质组研究等“规模型大科学”,并不属于经典的实验科学。这类工作中并不需要干涉,其目标只是把系统的全部元素测定清楚,以便得到一个含有所有信息的数据库。莱诺伊·胡德把这种类型的研究称为“发现的科学” (Discovery Science),而把上述依赖于干涉的实验科学称为“假设驱动的科学” (Hypothesis-driven science),因为选择干涉就是在做出假设。
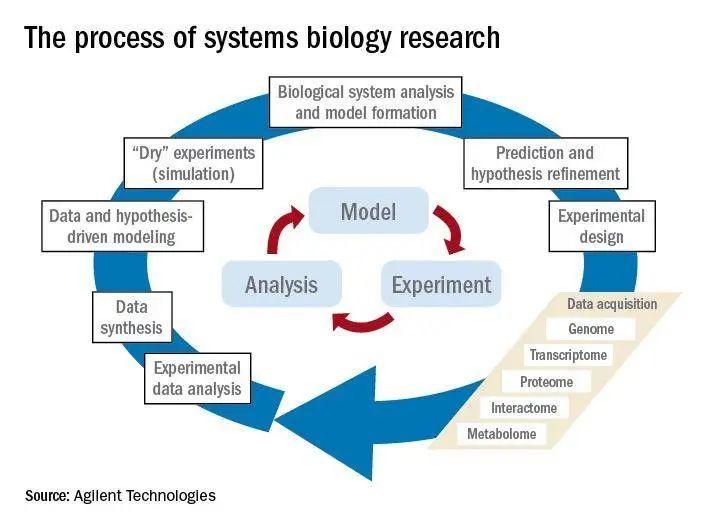
离开了数学和计算机科学,就不会有系统生物学。也许正是基于这一考虑,科学家把系统生物学分为“湿”的实验部分(实验室内的研究)和“干”的实验部分(计算机模拟和理论分析)。
图5:系统生物学包括“干”“湿”结合以不断优化生物模型的研究过程
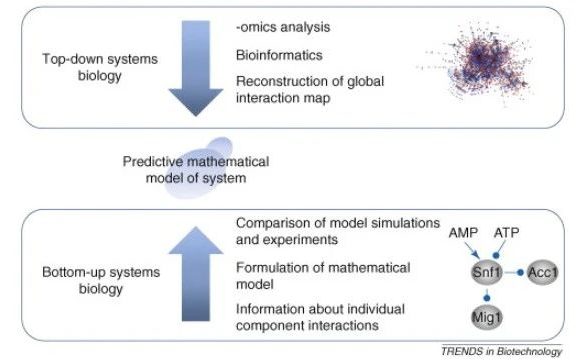
系统生物学的目的是在计算机模型(或数学模型)中重建一个生物系统,然后用来模拟系统的行为。基本上有两种系统生物学的方法,“自顶向下”从全部组学分析开始,而“自下而上”是从已知的单个路径开始,在实际应用中它们可以互补而不是相互排斥[4]。
图6:显示不同的系统生物学手段:在自顶向下的系统生物学中,高通量的实验数据,这些数据被用来重建通路或共调控模块。这些模块可以成为更详细的研究的基础,其中单个组件的动力学被量化。在自下而上的系统生物学中,通路的重建是基于对个体成分相互作用的研究。
自顶向下的系统生物学(Top-down systems biology)依赖于不同的全基因组分析,在这种分析中,数据是从一个暴露在不同条件下或受到基因干扰(如通过敲除特定基因)的系统中收集的。首先,基因、蛋白质和代谢产物表现出显著变化识别使用适当的统计方法(通常与多个校正测试),紧随其后的是集群和更高级的分析,得到的数据可以结合结构信息,例如 DNA 的蛋白质相互作用数据(从CHIP-chip实验)。这种类型的分析通常使识别协同调节模块成为可能。自顶向下的系统生物学的优点是它是非教条的,并且不需要通路结构的先验知识。由于假阳性的可能性,重构的通路需要用传统的分子生物学进行实验验证。
自下而上的系统生物学(Bottom-up systems biology)依赖于已知路径或子系统的可用知识。这种知识被组合成一个所谓的描述性模型的,并转换成数学模型。该数学模型可用于模拟不同条件下的路径运行。通过与实验数据的比较,有可能估计系统或路径内的个别过程的详细动力学。在得到系统的可接受的数学表示之前,常常需要对模型进行修正;因此,需要与实验工作密切配合,进行模型构建和仿真。所得模型可用于设计实验,进一步验证或证伪模型。这个过程的最终结果是一个动态数学模型,可以用来模拟所研究的生物系统。这种方法的缺点是,重建的模型高度依赖于当前已知系统通路的知识,尚未被识别成分的影响往往一开始完全被忽略掉。
以下推荐几本教材面向的受众是不同的,第一本适合入门,第二本和第三本则偏重哲学层面,第四本和第五本则更加偏向技术性。英文教材只有第五本有中译版。部分英文书籍介绍里附有链接。
-
-
《Systems Biology: Philosophical Foundations》
-
《Life: An Introduction to Complex Systems Biology》
-
《Systems Biology: Properties of Reconstructed Networks 》
-
《An Introduction to Systems Biology: Design Principles of Biological Circuits》
1. 张自立编著的《系统生物学》是目前我国高校使用的教材。整合了各层面的生物信息数据,建立各种数学模型进行仿真实验,进而定量阐明和预测生物功能、表型及行为。概述了系统生物学的基本概念和基本内容,介绍了基因组学(Genomics)、转录组学(Transcriptome)、蛋白质组学(Proteomics)、糖组学(Metabolomics)、代谢物组学(Metabolomics)、相互作用组学、表型组学、数学建模与仿真、序列比对与数据库搜索、分子进化模型与系统树的构建等。
2.《Systems Biology: Philosophical Foundations》是第一本关于系统生物学哲学基础的书。代表了近年来在与系统生物学相关的一系列哲学问题上的研究成果。包含十四篇论文,分为三个部分。第一部分描述了系统生物学研究计划(第2章和第5章),第二部分讨论了理论和模型(第6章和第9章),第三部分讨论了生物系统中的组织(第10章和第13章)。
https://www.sciencedirect.com/science/article/pii/B9780444520852500036?via%3Dihub#cesec1
图7:《Systems Biology: Philosophical Foundations》封面
3.《Life: An Introduction to Complex Systems Biology》这本 2003 年由 kaneko 写作的书,2006 年再版,本书一共有十二个章节,主要是面向年轻的生物学家和理论物理学家。这本提出的问题:什么是生命系统的通用属性和一个人怎么从生命的的现象学理论构造导致自然生殖细胞等复杂过程系统、进化和分化?
第一章,回顾了分子生物学的研究现状。对现状提出批评,以及需要的是另一种方法,作者称之为复杂系统生物学。在第2章中,作者概述了建构生物学的方法,即通过实验(在实验室中)和理论(在计算机模型中)构建生命的基本特征来理解它们。在第3章中,一些动力学系统和统计物理的基本背景被描述,作为在后面章节描述的研究的基础。第4章至第11章中讨论生命系统的基本问题。这些问题包括遗传(第4章)、繁殖和新陈代谢(第5章和第6章)、细胞分化、发育和形态发生(第7章和第9章)、与生物可塑性有关的进化(第10章)和多样化的物种形成(第11章)。在第12章中,总结了书中提出的基本概念,如稳定增长系统中的普遍统计、相应的多样化、合并、流动和少数控制原则。然后讨论如何理解生物的可塑性、递归性和进化性,同时强调现象学理论在生物学系统层面的必要性和可能性。
https://link.springer.com/book/10.1007/978-3-540-32667-0
图8:Kaneko《Life:An Introduction to Complex Systems Biology》封面
4. Palsson的《Systems Biology: Properties of Reconstructed Networks 》是更多从技术层面出发,以数学的方式表示化学计量矩阵,而这个矩阵的性质是决定它所代表的生化反应网络的官能状态的关键。这本教科书,致力于描述如何建模网络,如何确定他们的性质,以及如何将这些与表型功能重建为详细的,和可预测电路模型的生物系统。一些线性代数和生物化学的知识在阅读之前必不可少。
5.《An Introduction to Systems Biology:Design Principles of Biological Circuits》首次对系统生物学研究工作的核心和细节进行深入阐述,为直观理解生物学中一般原理建立了基础。Alon 的书适合假定没有生物学的知识,甚至对生物学不感兴趣的物理学家。全书内容编排:对网络模体等新的理论研究成果做了细致深入的阐述,指出了系统生物学的核心内容和实践方法;阐明了转录调控、信号转导、发育网络中的基本回路;检测了鲁棒性原理;清晰地说明了如何用进化优化来理解最优回路设计;仔细考虑了动力学校正和其他机制是如何使生物信息处理中的误差减到最少。
路德维希·冯·贝塔朗菲(Ludwig Von Bertalanffy,1901-1972)—美籍奥地利理论生物学家和哲学家;一般系统论的创始人,他从生物学领域出发,涉猎医学、心理学、行为科学、历史学、哲学等诸多学科,以其渊博的知识、浓厚的人文科学修养,创立了本世纪具有深远意义的一般系统论,使他的名字永久地与系统理论联系在一起。1972年,法国科学家委员会曾提名他为诺贝尔奖候选人,但是在诺贝尔奖评选委员会讨论提名之前,贝塔朗菲不幸辞世。
1950年发表《物理学与生物学中的开放系统理论》创立一般系统论并奠定了系统生物学的基础[5] [6]。我们可以看到,系统科学的研究和创立一开始就是和生物学息息相关的。
The Theory of Open Systems in Physics and Biology
https://science.sciencemag.org/content/111/2872/23/tab-pdf
An Outline of General System Theory (1950)
http://www.isnature.org/Events/2009/Summer/r/Bertalanffy1950-GST_Outline_SELECT.pdf
普利高津(Ilya Prigogine, 1917-2003)认为,只有在非平衡系统中,在与外界有着物质与能量的交换的情况下,系统内各要素存在复杂的非线性相干效应时才可能产生自组织现象,并且把这种条件下生成的自组织有序态称之为耗散结构。一个对象要想在活动中获得存在与发展,必须不断地从外界引入负熵,以抵消系统体内正熵的增加,从而确保自身不断地走向更高层次的稳定有序结构[7]。普利高津因此在 1977 获得诺贝尔化学奖。
Thermodynamic Theory of Structure, Stability and Fluctuations.
https://onlinelibrary.wiley.com/doi/abs/10.1002/bbpc.19720760520
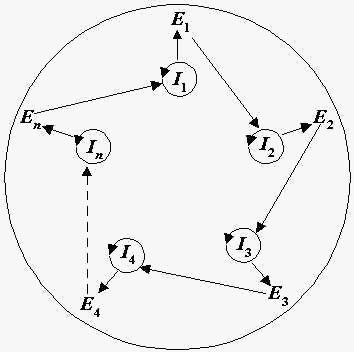
艾根(Manfred Eigen,1927-2019),1967年获得诺贝尔化学奖。超循环是一种自然自组织的原理,允许一组功能耦合的自代表实体的连续一致演化。超循环是一类新颖的非线性反应网络,它是一个能够自我指导自身复制的整体,并为下一个循环的复制提供了催化支持[8] [9]。如下图所示,信息载体 I 不仅包含了自身复制的信息,还包含了具有促使转化成其他类型所对应功能特性的媒介物 E(通常是一种酶)的信息。这种由载体信息生成的酶,支持了下一个信息载体的活性。一个超催化循环由若干网状的催化循环形成,必须的两种功能是,每个循环能够自我复制,并且一个循环的产物必须支持下一个循环。
图9:超循环是一种自我复制的大分子系统,其中 RNA(I)和酶(E)的协同作用。
Self organization of matter and the evolution of biological macromolecules
https://link.springer.com/article/10.1007/BF00623322
The Hypercycle: A principle of natural self-organization
https://onlinelibrary.wiley.com/doi/abs/10.1002/qua.560140722
作为人类基因组计划的发起人之一,美国科学家莱诺伊·胡德(Leroy Hood)也是组学 (Omics) 生物技术开创者之一。胡德已经开发了突破性的科学仪器,使生物科学和医学科学的重大进展成为可能。这些包括用于确定组成给定蛋白质的氨基酸的第一气相蛋白质测序器,DNA 合成器,用来合成 DNA 的短片段,肽合成器,将氨基酸结合成较长的肽和较短的蛋白质,第一个自动DNA测序器[10] [11]。
https://www.ncbi.nlm.nih.gov/pubmed/12540920
Systems Biology and New Technologies Enable Predictive and Preventative Medicine
https://api.semanticscholar.org/CorpusID:33015388
1996 年在北京举办的第 1 届国际转基因动物学术研讨会,中科院曾邦哲阐述了系统论与生物遗传学、转基因研究等,1999 年元月于德国建立了系统生物科学与工程网(http://www.sysbioeng.com/genozen/project.html)表述生物系统结构论(Structurity theory)的结构整合 (Integrative)、调适稳态(Stability)与层级建构(Constructive) 等综合(Synthetic)系统理论规律,并定义实验、计算系统研究,同系统科学、计算机科学、纳米科学和生物医学、生物工程等领域国际科学家广泛通讯,倡导分子生物技术和计算机科学 -实验生物学家与计算生物学家结合研究生物系统,唤起了一大批生物学研究领域以外的专家的关注。
1999 年更早的中期不少科学家开始了论述,2000 年日本举办了国际系统生物学会议,随后,系统生物学便逐渐重新得到了生物科学界的认同。2002 年日本北野宏明(Kitano H.)也论述了系统生物学是实验与计算方法整合的生物系统研究[12] [13]。
Computational systems biology
https://doi.org/10.1038/nature01254
The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models
https://doi.org/10.1093/bioinformatics/btg015
2004 的综述里阐述了生物系统的鲁棒性:鲁棒性是生物系统普遍存在的特性。它被认为是复杂演化系统的一个基本特征。它是通过对生物有机体和复杂工程系统普遍适用的几个基本原则来实现的。鲁棒性促进了可进化性,而鲁棒性特征通常是由进化选择的[14]。
https://www.ncbi.nlm.nih.gov/pubmed/15520792
2000 年美国 E. Kool 基于系统生物学的基因工程,重新提出合成生物学(Synthetic biology)。
合成生物学是一门将科学与工程相结合,以设计和构建新的生物功能和系统的生物学研究新领域。合成生物学的定义已被普遍接受为生物学工程:综合复杂的、基于生物学的(或启发的)系统,这些系统显示了自然界中不存在的功能。这种工程学的观点可以应用于生物结构的各个层次,从单个分子到整个细胞、组织和生物体[15]。
Synthetic Biology, Volume 1
https://www.overdrive.com/media/2502000/synthetic-biology-volume-1
合成生物学家分为两大类。一种使用非自然分子来重现自然生物学中出现的行为,目的是创造人工生命。另一种则从自然生物学中寻找可互换的部分,将其组合成非自然功能运行的系统[16]。
http://dx.doi.org/10.1038/nrg1637
2008 年 Nature 文章则论述了系统生物学与合成生物学的结构理论[17]。
Systems and Synthetic biology: tackling genetic networks and complex diseases
https://www.nature.com/articles/hdy200918
生物信息学(Bioinformatics)包括开发和应用软件工具,以帮助理解生物功能和数据,而系统生物学涉及数学和计算建模的生物系统和功能,以简化表示,理解和文档。生物信息学整合和应用统计学、数学、计算机科学、工程和生物学的理论和实践知识,并允许在生物数据的电脑分析和计算机化解释的数据。另一方面,系统生物学利用信号通路、代谢网络和基因序列功能的知识,以促进科学的研究和应用。
生物信息学最早出现在 50 多年前,当时台式电脑还只是一种假设,DNA 还无法测序。20 世纪 60 年代,第一个新的肽序列组装器、第一个蛋白质序列数据库和第一个用于系统发育的氨基酸替代模型被开发出来。在 20 世纪 70 年代和 80 年代,分子生物学和计算机科学的并行发展为分析全基因组等日益复杂的工作铺平了道路。在 1990 年的 21 世纪头十年,互联网的使用,加上二代测序,导致了数据的指数增长和生物信息学工具的迅速发展。今天,生物信息学面临着多种挑战,如处理大数据、确保结果的再现性以及与学术领域的恰当交融[18]。
玛格丽特·达霍夫(Margaret Dayhoff,1925-1983)是一位美国物理化学家,她率先将计算机方法应用于生物化学领域[19]。
Digital electronic computers in biomedical science.
https://www.ncbi.nlm.nih.gov/pubmed/14415153

图10:NCBI 基因库和 WGS(Whole Genome Shotgun,全基因组鸟枪法)数据库中随时间变化的序列总数。
在 1990 年代到 2000 年代,测序技术的重大改进以及成本的降低使数据呈指数级增长。自 2008 年以来,摩尔定律不再是 DNA 测序成本的准确预测指标,在大规模并行测序技术出现后,摩尔定律降低了几个数量级。
虽然在某些情况下,根据必要的计算,一台简单的台式计算机就足够了,但生物信息学的一些项目将需要更庞大、昂贵和需要专门知识的基础设施。一些政府资助的专门从事高性能计算的组织已经出现,例如:
-
Compute Canada (https://www.computecanada.ca)
-
New York State’s High Performance Computing Program
(https://esd.ny.gov/new-york-state-high-performance
-
The European Technology Platform for High Performance Computing (http://www.etp4hpc.eu/)
-
China’s National Center for High-Performance Computing (http://www.nchc.org.tw/en/)
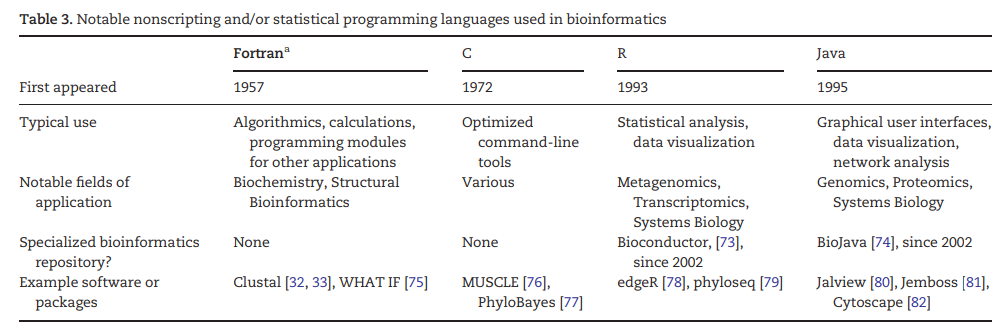
图11:一些生物信息学中使用的非脚本和/或统计编程语言,以及应用软件和程序包[18]。
2002 年03 月,美国《Science》周刊登载了系统生物学专集(链接:http://science.sciencemag.org/content/295/5560),该专集导论中的第一句话这样写道:“如果对当前流行的、前沿的关键词进行一番分析,那么人们会发现,‘系统’高居在排行榜上。”专题中包含四篇综述论文:
图12:专题封面图:以模块化闻名的乐高积木,是对生物架构和动态过程一个恰当的比喻,包括从基因表达到组织和有机体的功能各个层次,组件之间的联系、如何被管理的,以及它们是如何进化的。这些都是理解不同层次生物复杂性的关键。
第一篇是有北野宏明所作,阐述了从系统的层面理解生物学,我们必须研究细胞和有机体功能的结构和动态,而不是细胞或有机体的孤立部分的特征。系统的特性,如鲁棒性,成为中心问题,了解这些特性可能会对医学的未来产生影响。然而,在系统生物学的成就能够发挥其备受吹捧的潜力之前,需要在实验装置、先进软件和分析方法上取得许多突破[20]。
Systems Biology: A Brief Overview
https://science.sciencemag.org/content/295/5560/1662
第二篇综述从工程理论和实践中阐明一些生物复杂性的见解。先进的技术和生物学有着截然不同的物理实现,但它们在系统级组织方面的相似之处远比人们普遍认为的要多。这两个领域的趋同演化产生了由协议的精细层次结构和反馈调节层组成的模块化架构,这些架构是由对不确定环境的鲁棒性需求驱动的,并且经常使用不精确的组件。这些令人困惑和矛盾的特征既不是偶然的,也不是人为的,而是源于复杂性和鲁棒性、模块化、反馈和脆弱性之间深刻而必要的相互作用[21]。
Reverse Engineering of Biological Complexity
https://science.sciencemag.org/content/295/5560/1664
第三篇以海胆为例,阐述了胚胎内胚层和中胚层规格的基因调控网络。该网络是由大规模扰动分析,结合计算方法,基因组数据,顺式调控分析,和分子胚胎学。该网络目前包含 40 多个基因,每个节点都可以通过顺式调控分析在 DNA 序列水平上直接验证。其结构体系揭示了发育的具体和一般方面,例如特定的细胞如何在胚胎中产生它们指定的命运,以及为什么这一过程在发育过程中不可阻挡地向前发展[22]。
A Genomic Regulatory Network for Development
https://science.sciencemag.org/content/295/5560/1669
第四篇综述以心脏这一器官为例,阐述了器官建模的进展。成功的生理分析需要理解细胞、器官和系统的关键组成部分之间的功能相互作用,以及这些相互作用在疾病状态中是如何变化的。这些信息既不存在于基因组中,也不存在于基因编码的单个蛋白质中。它存在于亚细胞、细胞、组织、器官和系统结构中蛋白质相互作用的水平。因此,除了复制自然和计算这些交互来确定健康和病理状态的逻辑之外,没有其他选择。生物数据库的迅速增长;细胞、组织和器官模型;而强大的计算硬件和算法的发展使得从基因到整个器官和调节系统的生理功能的定量探索功能成为可能[23]。
Modeling the Heart–from Genes to Cells to the Whole Organ
https://science.sciencemag.org/content/295/5560/1678
根据使用跨学科工具从多个实验中获得,整合和分析复杂数据集的能力的系统生物学解释,一些典型的技术平台包括:
基因组学,表观遗传组,转录组学,蛋白质组学,代谢物组学,糖组学,脂类组学(Lipomics),除了上述给定分子的识别和量化之外,进一步的技术还分析细胞内的动力学和相互作用。包括:相互作用组学(Interactomics),代谢流组学(Fluxomics),生物组学(Biomics)。
其他技术如计算机科学,信息学和统计学的其他方面也用于系统生物学。
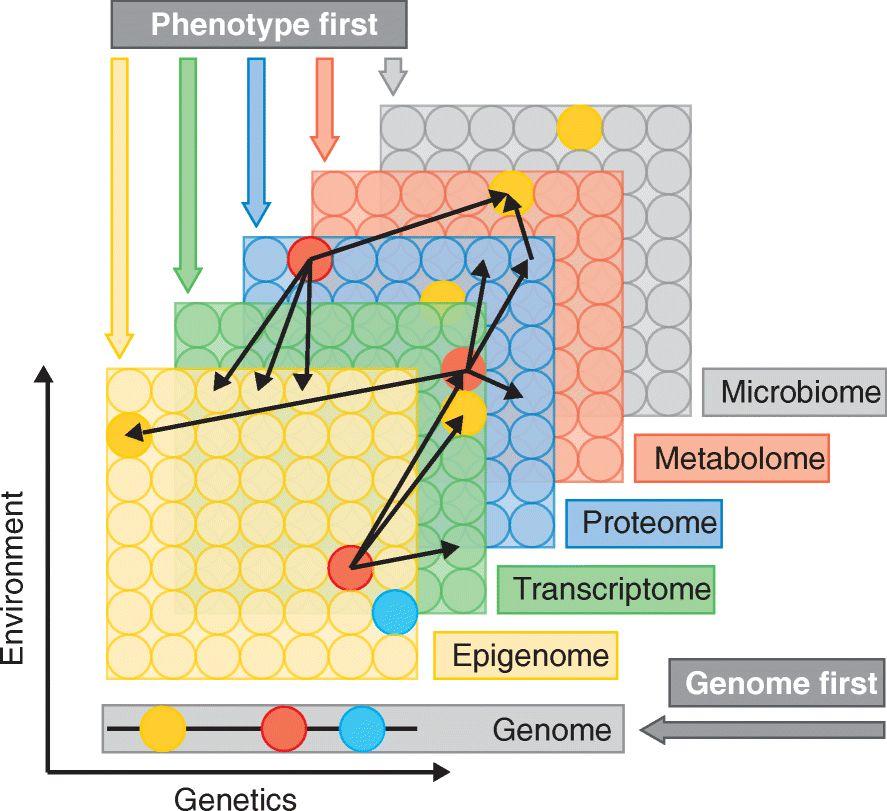
图13:多组学方法对某种特定疾病的研究。组学数据收集在整个分子池上,以圆圈表示。除了基因组外,所有的数据层都同时反映了遗传调控和环境,这可能会对每个个体分子产生不同程度的影响。细黑箭头表示在不同层中检测到的分子之间潜在的相互作用或相关性。例如,红色的转录本可以与多种蛋白质相关联。在同一层中,交互也很普遍,图中未表述[24]。
为了获得全面、独立、高质量的中国人群特异性基因组数据库,中国代谢解析计划ChinaMAP(China Metabolic Analytics Project)诞生了。4月30日,ChinaMAP在《Cell Research》杂志上发表了一期研究成果,首次报道了来自全国 27 个省份和直辖市、8个民族超过一万人的深度全基因组测序数据分析,发现了 1.36 亿个单核苷酸多态性(SNPs)和 1070 万个插入或缺失位点(INDEL),其中一半以上是未在其他数据库报道过的新突变[25]。
The ChinaMAP analytics of deep whole genome sequences in 10,588 individuals
https://doi.org/10.1038/s41422-020-0322-9
该文系首次正式发表大规模东亚南北方史前人类基因组分析结果,为探源华夏族群及其文化和修正东亚南方人群演化模式做出了重大贡献。在中华民族探源方面,发现中国、东亚主体人群连续演化是主旋律,中国南北方古人群早在 9500 年前已经分化,至少在 8300 年前南北人群融合与文化交流的进程即已开始,4800 年前出现强化趋势,至今仍在延续[26]。
American Association for the Advancement of Science
https://www.britannica.com/topic/American-Association-for-the-Advancement-of-Science
泛癌症全基因组(Pan-cancer Genomics)
Nature 杂志2020年2月份整理了数篇全基因组的泛癌症分析的文章(链接:https://www.nature.com/collections/afdejfafdb)。
癌症是一种基因组疾病,由细胞获得关键癌症基因的体细胞突变引起。这些突变改变了调节细胞生长和与组织环境相互作用的途径。直到最近,对癌症基因组的研究都集中在蛋白质编码基因上,这些基因加起来只占基因组的 1%。为了解决这个问题, ICGC/TCGA 全基因组癌症分析(PCAWG)项目对超过 2600 种原发癌症及其 38 种不同肿瘤类型的正常组织进行了全基因组测序和综合分析。这项研究揭示了广泛的大规模结构性突变在癌症所扮演的角色,确定这种癌症相关的突变基因调控区域,推测肿瘤进化多个癌症类型,照亮了体细胞突变和转录组之间的相互作用和研究生殖系遗传变异的作用在调节突变过程[27] [28] [29]。
Pan-cancer analysis of whole genomes
https://www.nature.com/articles/s41586-020-1969-6
Patterns of somatic structural variation in human cancer genomes
https://doi.org/10.1038/s41586-019-1913-9
Analyses of non-coding somatic drivers in 2,658 cancer whole genomes
https://doi.org/10.1038/s41586-020-1965-x
2020年5月11日,哈佛医学院的 Andrew D. Cherniack 和 Rameen Beroukhim 合作发表文章Comprehensive Analysis of Genetic Ancestry and Its Molecular Correlates in Cancer,通过分析 TCGA 数据库的 33 种癌症类型的 10678 名病患的突变速率、DNA甲基化、mRNA 和 miRNA 表达,鉴定出与癌症相关的遗传性祖先因素(Ancestry effect)[30]。
Comprehensive Analysis of Genetic Ancestry and Its Molecular Correlates in Cancer, Cancer Genome Atlas Analysis Network
https://pubmed.ncbi.nlm.nih.gov/32396860/
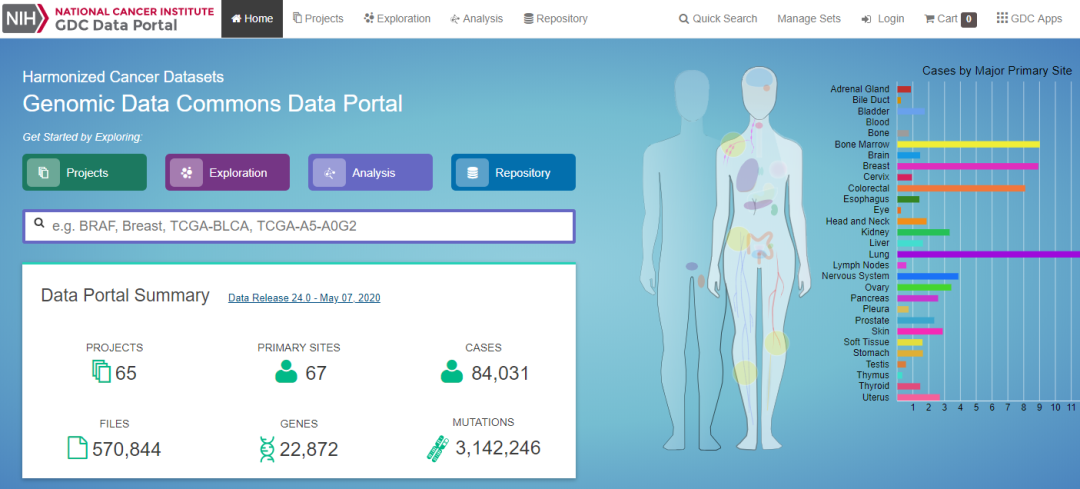
TCGA (The Cancer Genome Atlas)数据库 https://portal.gdc.cancer.gov/ ,癌症基因组图谱(TCGA)是一个具有里程碑意义的癌症基因组学项目,它对 20000 多例原发性癌症进行了分子特征分析,并对 33 种癌症类型的正常样本进行了匹配。国家癌症研究所和国家人类基因组研究所的这项联合工程始于 2006 年,汇集了来自不同学科和不同机构的研究人员。
图14:TCGA 数据库首页,可以根据感兴趣的癌症或者基因名进行检索分析
癌症系统生物学的一个关键目标是利用大数据来阐明癌症发生的分子网络。但是,到目前为止,还没有系统地评价这些努力取得了多大进展。下文中作者调查了六种主要的系统生物学方法,以绘制和建模癌症路径,并注意到他们的网络地图覆盖和增强现有的知识[31]。
A census of pathway maps in cancer systems biology
https://doi.org/10.1038/s41568-020-0240-7
2019年7月10日,来自美国 NIH 的 Ananda L. Roy 团队回顾了 NIH 表观遗传组学蓝图计划(Roadmap)启动的契机和总体目标;介绍了表观遗传组学项目的成果:参考表观遗传组、国际间表观遗传的合作研究、疾病的表观遗传基础和新型表观遗传标志物的发现、表观遗传研究技术的发展等;总结了项目实行过程中的经验和教训[32]。
The NIH Common Fund/Roadmap Epigenomics Program: Successes of a comprehensive consortium
https://doi.org/10.1126/sciadv.aaw6507
人类 SRMAtlas:代表人类蛋白质组的 166174 个蛋白型肽,提供了多种独立的分析方法来量化任何人类蛋白和大量的剪接变异、非同义突变和翻译后修饰[33]。本文通讯作者为 Hood L。
Human SRMAtlas:A Resource of Targeted Assays to Quantify the Complete Human Proteome
https://www.sciencedirect.com/science/article/pii/S0092867416308492
2020年4月,Nature 发表了一篇文章介绍了一个是一种系统的全蛋白质参考平台:HuRI,它将基因组变异与表型结果联系起来。一个包含约 53000 个蛋白质与 8000 多个蛋白质相互作用的人类二元蛋白质相互作用图,为研究健康和疾病中的人类细胞功能提供了参考。推测的组织特异性网络揭示了细胞环境特异性功能形成的一般原则,并阐明可能构成孟德尔疾病组织特异性表型的潜在分子机制[34]。
A reference map of the human binary protein interactome
https://www.nature.com/articles/s41586-020-2188-x
许多栖息在人体的微生物与人类的健康和疾病密切相关,但大部分微生物对我们而言仍然是未知的。美国国立卫生研究院(NIH)人体微生物组项目的研究人员已经确定,仅人体肠道微生物组就有 100 万亿细菌,是人体细胞数量的 10 倍。此外,它还含有大约 800 万个蛋白质编码基因,是人类基因组的 360 倍。科学家们已经了解到,这些细菌组成的变化——生态系统中的一种干扰——可能与一系列人类疾病有关,包括炎症性肠病、哮喘、关节炎和多发性硬化症[35]。
图15:继人类基因组计划之后,美国国立卫生研究院在2007年启动了一个类似的雄心勃勃的计划——人类微生物组计划(Human Microbiome Project,HMP),https://www.hmpdacc.org/。
https://science.sciencemag.org/content/336/6086/1209
意大利特伦托大学的一支研究团队展开一项超大规模研究,样本涵盖了不同地理位置、年龄和生活方式的人群以及人体的不同部位。他们利用单样本宏基因组组装,构建出超过 15 万个人体微生物基因组,其中 77% 以前从未被描述过,确定了一些普遍存在、但以前未被发现的微生物类群[36]。
Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle
https://doi.org/10.1016/j.cell.2019.01.001
作为 ICGC/TCGA 全基因组泛癌分析(PCAWG)联盟的一部分,作者训练了一个深度学习分类器,以基于全基因组测序(WGS)中检测到的代表 PCAWG 联盟产生的 24 种常见癌症类型的 2606 种肿瘤的体细胞旅客突变模式来预测癌症类型。分类器在切除肿瘤样本上的准确率分别为 91%,在独立原发和转移样本上的准确率分别为 88% 和 83%,大约是训练过的病理学家在不了解原发样本的情况下对转移肿瘤的准确率的两倍[37]。
A deep learning system accurately classifies primary and metastatic cancers using passenger mutation patterns
https://www.nature.com/articles/s41467-019-13825-8
理解复杂的生物系统需要软件工具的广泛支持。系统生物学计算工作流程的每一步都需要这些工具,通常包括数据处理、网络推理、深度筛选、动态模拟和模型分析。此外,现在正在努力开发集成的软件平台,以便在工作流程的不同阶段以及由不同的研究人员使用的工具可以很容易地一起使用。这篇综述描述了在系统生物学研究的不同阶段所需要的软件工具的类型,以及目前可供系统生物学研究人员使用的选择[38]。
Software for systems biology: from tools to integrated platforms
https://doi.org/10.1038/nrg3096
[1] Cancer Cell.2002 Sep;2(3):179-82. doi: 10.1016/s1535-6108(02)00133-2
[2] Karr, Jonathan R. et al. “A Whole-Cell Computational Model Predicts Phenotype from Genotype.” Cell 150 (2012): 389-401.
[3] Feist, Adam M. et al. “Reconstruction of biochemical networks in microorganisms.” Nature Reviews Microbiology 7 (2009): 129-143.
[4] Petranovic, Dina and Jens Nielsen. “Can yeast systems biology contribute to the understanding of human disease?” Trends in biotechnology 26 11 (2008): 584-90 .
[5] Science 13 Jan 1950:Vol. 111, Issue 2872, pp. 23-29
[6] Ludwig von Bertalanffy, “An Outline of General System Theory,” The British Journal for the Philosophy of Science, Vol. 1, No. 2 (Aug., 1950), pp. 134-165.
[7] P. Glansdorff und I. Prigogine: Thermodynamic Theory of Structure, Stability and Fluctuations , Wiley‐Interscience, London 1971,306 S.
[8] Eigen, M. Self-organization of matter and the evolution of biological macromolecules. Naturwissenschaften 58, 465–523 (1971).
[9] Eigen, Manfred and Peter Schuster. “The Hypercycle: A principle of natural self-organization.” (1979).
[10] Hood, L., & Galas, D.J. (2003). The digital code of DNA. Nature, 421, 444-448.
[11] Hood, L., Heath, J.R., Phelps, M.E., & Lin, B. (2004). Systems biology and new technologies enable predictive and preventative medicine. Science, 306 5696, 640-3 .
[12] Kitano, H. (2002). Computational systems biology. Nature, 420, 206-210.
[13] Hucka, Michael et al. “The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models.” Bioinformatics 19 4 (2003): 524-31 .
[14] Kitano, H. (2004). Biological robustness. Nature Reviews Genetics, 5, 826-837.
[15] https://www.overdrive.com/media/2502000/synthetic-biology-volume-1
[16] Benner SA, Sismour AM. Synthetic biology. Nat Rev Genet. 2005;6(7):533‐543. doi:10.1038/nrg1637
[17] Cuccato G, Della Gatta G, di Bernardo D. Systems and Synthetic biology: tackling genetic networks and complex diseases. Heredity (Edinb). 2009;102(6):527‐532. doi:10.1038/hdy.2009.18
[18] Gauthier, J., Vincent, A.T., Charette, S.J., & Derôme, N. (2019). A brief history of bioinformatics. Briefings in bioinformatics.
[19] Ledley, Robert S.. “Digital electronic computers in biomedical science.” Science 130 3384 (1959): 1225-34 .
[20] Kitano, H. (2002). Systems biology: a brief overview. Science, 295 5560, 1662-4 .
[21 Csete, M., & Doyle, J.C. (2002). Reverse engineering of biological complexity. Science, 295 5560, 1664-9 .
[22] Davidson, Eric H. et al. “A genomic regulatory network for development.” Science 295 5560 (2002): 1669-78 .
[23] Noble, D. (2002). Modeling the heart–from genes to cells to the whole organ. Science, 295 5560, 1678-82 .
[24] Hasin, Yehudit et al. “Multi-omics approaches to disease.” Genome Biology 18 (2017): n. pag.
[25] Cao, Yanan et al. “The ChinaMAP analytics of deep whole genome sequences in 10,588 individuals.” Cell Research (2020): 1 – 15.
[26] [The Editors of Encyclopaedia Britannica](https://www.
britannica.com/editor/The-Editors-of-Encyclopaedia-Britannica/4419)
[27] PeterJ.GadJanO.JoshuaM.JenniferL.LincolnD. et al. “Pan-cancer analysis of whole genomes.” Nature 578 (2020): 82 – 93.
[28] Li, Yilong et al. “Patterns of somatic structural variation in human cancer genomes.” Nature 578 (2020): 112 – 121.
[29] Rheinbay, Esther et al. “Analyses of non-coding somatic drivers in 2,658 cancer whole genomes.” Nature 578 (2020): 102 – 111.
[30] Carrot-Zhang J, Chambwe N, Damrauer JS, et al. Comprehensive Analysis of Genetic Ancestry and Its Molecular Correlates in Cancer. Cancer Cell. 2020;37(5):639‐654.e6. doi:10.1016/j.ccell.2020.04.012
[31] Kuenzi, B.M., & Ideker, T. (2020). A census of pathway maps in cancer systems biology. Nature Reviews Cancer, 20, 233 – 246.
[32] Satterlee, John S. et al. “The NIH Common Fund/Roadmap Epigenomics Program: Successes of a comprehensive consortium.” Science Advances 5 (2019): n. pag.
[33] Kusebauch, Ulrike et al. “Human SRMAtlas: A Resource of Targeted Assays to Quantify the Complete Human Proteome.” Cell 166 (2016): 766-778.
[34] Luck, Katja et al. “A reference map of the human binary protein interactome.” Nature580 (2020): 402 – 408.
[35]:Hood, L. (2012). Tackling the microbiome. Science, 336 6086, 1209 .
[36] Pasolli, Edoardo et al. “Extensive Unexplored Human Microbiome Diversity Revealed by Over 150,000 Genomes from Metagenomes Spanning Age, Geography, and Lifestyle.” Cell 176 (2019): 649 – 662.e20.
[37] Jiao, Wei et al. “A deep learning system accurately classifies primary and metastatic cancers using passenger mutation patterns.” Nature Communications 11 (2020): n. pag.
[38] Ghosh, Samik et al. “Software for systems biology: from tools to integrated platforms.” Nature Reviews Genetics 12 (2011): 821-832.
(参考文献可上下滑动)
生物网络 | 集智百科
进化新视角:基因之间的重复博弈如何影响生物进化?
Physics Reports计算网络生物学长文综述:数据、模型和应用
意识如何自下而上地涌现?来自系统生物学的启示 | 长文综述
加入集智,一起复杂!

集智俱乐部QQ群|877391004
商务合作及投稿转载|swarma@swarma.org
搜索公众号:集智俱乐部
加入“没有围墙的研究所”
让苹果砸得更猛烈些吧!
👇点击“阅读原文”,了解更多论文信息