基于计算学方法的蛋白质相互作用预测综述

导语
今天给大家介绍来自中科院的胡伦和IBM的胡鹏伟等人在Briefings in Bioinformatics上发表的文章“A survey on computational models for predicting protein-protein interactions”。预测蛋白质之间的相互作用(PPI)对研究生物体内的各种细胞学机制至关重要,计算学方法能够有效改善传统生物学方法预测PPI时耗时耗力,且预测结果不可靠的问题。在本文中,我们描述了PPI预测所需的各种蛋白质相关数据库,介绍了现有的各种计算学模型的优缺点,然后描述了常用的实验方案和模型性能评价指标,并介绍了几种在线预测工具,最后阐明了预测PPI的未来发展方向。

DrugAI | 来源

论文标题:
A survey on computational models for predicting protein-protein interactions
论文地址:
https://academic.oup.com/bib/advance-article-abstract/doi/10.1093/bib/bbab036/6159365?redirectedFrom=fulltext
简介
简介
作为细胞中最常见的分子之一,蛋白质对于调节细胞中的各种新陈代谢途径以及众多生物学过程具有十分重要的意义。一般来说,蛋白质并不是单独发挥作用的,而是通过彼此之间发生相互作用,即蛋白质-蛋白质相互作用(PPI)来完成相应的任务。除此以外,对蛋白质相互作用的研究能够为医学诊断和治疗提供新视角,促进新药的设计以及生物医学的发展。因此,预测PPI已成为系统生物学的基础课题,且引起了越来越多的关注。
目前,预测蛋白质相互作用的方法主要包括生物学方法和计算学方法两种,在传统的生物学领域,相互作用数据的收集可通过酵母双杂交、蛋白质芯片、合成致死分析等方法完成,然而,这些方法既耗时又费力,导致预测效率不足,且预测结果中经常能观察到该比例的假阴性和假阳性现象。因此,随着计算机技术的高速发展,原本作为辅助手段的计算学方法,目前已经成为预测蛋白质相互作用的主流方法。
计算学预测模型可根据使用预测信息的不同被分为以下五种:基于网络结构的模型、基于序列的模型、基于结构的模型、基于基因组的模型、基于基因本体论的模型。其中,第一种模型利用给定的蛋白质相互作用网络,从网络结构中挖掘不同的信息,设计不同的拓扑相似度度量方法,根据已知的相互作用预测未知的相互作用。后四种模型利用蛋白质中的各种生物学信息,如:蛋白质序列、结构、基因组、基因本体论等提取能为相互作用预测提供帮助的数据,为蛋白质对构建特征向量,再结合分类器完成预测任务。除此以外,结合发展迅速的深度学习技术以及MapReduce技术等,计算学模型还发展出了另外两个分支:基于深度学习的模型和大规模预测模型。
在本文中,回顾了上述七种模型预测蛋白质相互作用的方法。
常用数据库
常用数据库
随着高通量技术的发展,已建立了许多与蛋白质有关的数据库,这些数据库包含了蛋白质不同的信息,是计算学预测蛋白质相互作用的重要资源。下面,我们将回顾几种常用的数据库,根据所包含的信息,这些数据库被分为五类:蛋白质相互作用网络、蛋白质序列、高级结构、基因组信息、基因本体论。
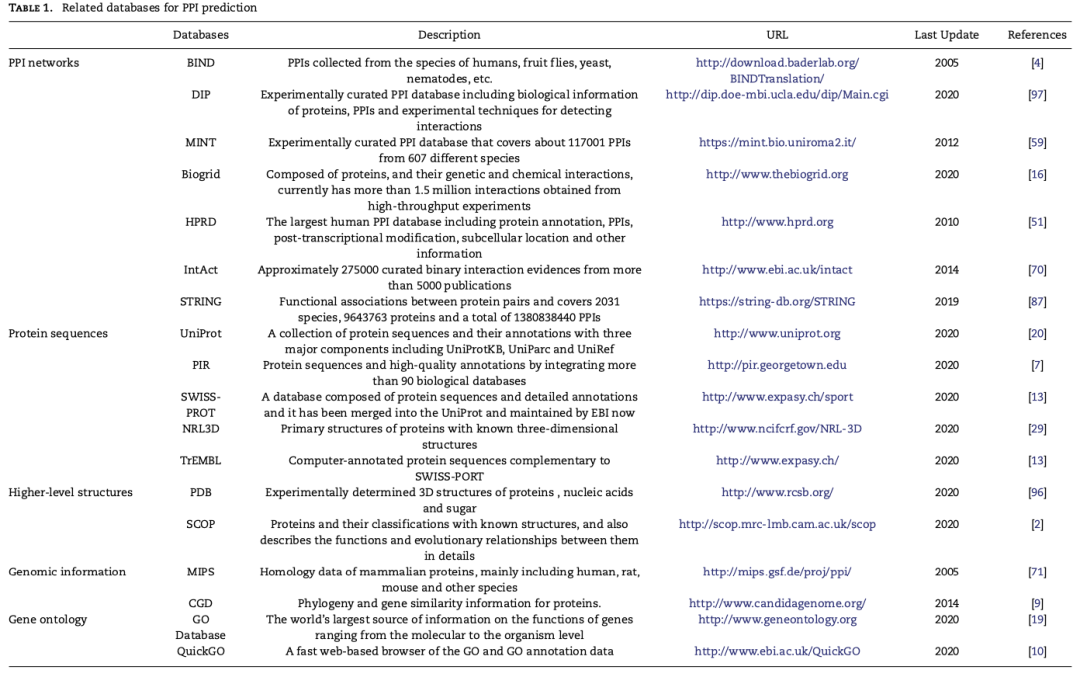
蛋白质相互作用网络数据库
蛋白质相互作用网络是由相互作用数据构建而来的,多种常见的数据库都能提供各个物种的相互作用网络信息,例如BIND、DIP、MINT、BioGRID、HPRD、IntAct和STRING。由于这些数据库提供的相互作用是通过生物学方法验证的,因此利用这种真实验证过的数据进行预测具有更高的准确性。此外,在这些数据库中,MINT、IntAct和STRING还提供了从不同来源获得的PPI分数,用来评估相互作用的可靠性。在实际应用时,也可以通过挑选得分较高的蛋白质对来构建更可靠的PPI网络。
蛋白质序列数据库
蛋白质序列也可被称作蛋白质一级结构,它指的是氨基酸残基在蛋白质肽链中的排列顺序,是蛋白质最基础的结构。相关蛋白质序列信息可从UniProt、PIR、SWISS-PROT、NRL3D和TrEMBL数据库获得,它们都包括了各种生物的蛋白质序列信息和相关注释信息。
高级结构数据库
除了上述的一级结构外,蛋白质还有二级、三级和四级三个更高级的结构,它们都是由一级结构决定的蛋白质的空间结构。在一级结构序列中,蛋白质肽链是直链状,而二级结构中的肽链分子会通过一定的规律进行卷曲或折叠形成的特定空间结构,如α螺旋和β折叠;三级结构是在二级结构的基础上进一步盘曲或折叠形成的三维(3D)空间结构;四级结构则是具有两条或两条以上三级结构的多肽链组成的蛋白质。其中最常被用于预测的是蛋白质的三维结构,该信息可以从PDB和SCOP获得。
基因组信息数据库
由于全基因组测序技术的高速发展,多种现象如:基因融合、基因邻接和系统发育图谱可以被很好的观察到,而这些信息已被多项研究证明可用于预测蛋白质间的相互作用。此类信息可在MIPS和CGD数据库中获得,前者更多的是哺乳动物相关的基因信息,后者包括其他多种生物。
基因本体论数据库
基因本体论(Gene Ontology,GO)是用于描述基因及其产物的功能和联系的,而蛋白质就是常见的基因产物,基因本体论包括三部分:细胞成分、分子功能和生物学过程。相关信息可以从GO database和QuickGO中下载。
计算学预测模型
计算学预测模型
我们将介绍计算学预测模型的七大种类,并分析它们的优缺点,包括:基于网络的模型、基于序列的模型、基于结构的模型、基于基因组的模型、基于基因本体论的模型、基于深度学习的模型和大规模预测模型。
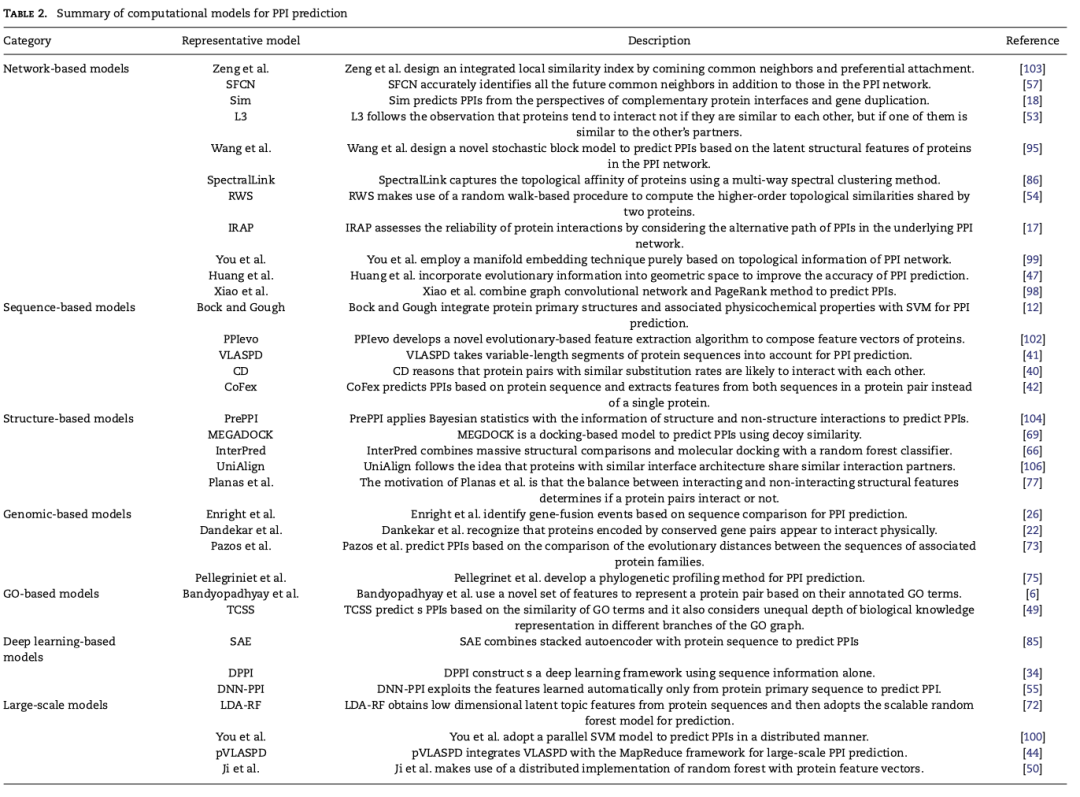

基于网络结构的模型
一般而言,基于网络结构的预测模型会把蛋白质相互作用网络看作图论中的无权无向图,记作,其中,是个蛋白质的集合,是中一个的邻接矩阵。给定两个蛋白质和,如果和具有相互作用,则,否则。
这类模型能够分析网络中潜在的结构信息,然后利用不同的结构信息和度量方法计算两个蛋白质之间的拓扑相似度,从而评估它们发生相互作用的可能性。常见的能够被用于预测相互作用的网络结构信息包括共同邻居、网络路径、全局网络结构和几何嵌入四种。这四类方法能够从局部和全局的角度衡量蛋白质对拓扑相似性,以获取更高的预测性能。
基于序列的模型
基于此信息的预测模型主要通过蛋白质序列提取某些能够为预测任务提供支持的信息,例如氨基酸的疏水性、亲水性等,然后利用这些信息为每个蛋白质生成唯一特定的特征向量,最后把提取出的蛋白质向量输入到经典的分类器中,如支持向量机(Support Vector Machine,SVM)和随机森林(Random Forests,RF),对蛋白质对进行二分类处理,由此获取预测结果。
此类模型能够基于序列从多种角度预测相互作用,如:序列相似性和共同进化信息,并通过不同的方法丰富预测信息,更准确的识别有用的蛋白质序列,进一步提升模型的预测性能。
基于结构的模型
除了序列外,蛋白质还有二级结构、三级结构和四级结构,这三个结构都是蛋白质在一级结构的基础上形成的更高维度也更复杂的空间结构。由于目前对这些高级结构的认知远不如蛋白质序列那么多,所以,基于此类信息预测蛋白质相互作用的模型数量也要大幅度小于基于序列的模型。目前,蛋白质的三级结构是最常见的被用于预测相互作用的高级结构信息,基于该信息进行预测的模型是根据以下假说工作的:如果两个蛋白质的相互作用区域能够完美嵌合,那么它们之间很可能存在相互作用。
基于基因组的模型
随着多种生物基因组测序工作的完成,很多现象如:基因融合、基因顺序和系统发育图谱等可以被更好地观察到,其中,基于基因融合的预测模型认为能够在另一个组织中彼此融合的两个蛋白质,更有可能彼此发生相互作用;基于基因顺序的模型认为基因之间的顺序表明了其在功能上的相关性,而功能相关的蛋白质对更可能发生相互作用;系统发育图谱通常存在于系统发育树中,基于该信息的模型认为蛋白质系统发育图谱的相似性表明了其在功能上的相关性,因此具有相似发育图谱的蛋白质更可能发生相互作用。虽然基于基因组的模型历史悠久且具有较好的性能,但是对于那些没有发现基因融合事件,基因顺序无法获取,以及不能很好的映射其系统发育图谱的蛋白质对,它们之间的相互作用无法利用此类模型进行预测。
基于基因本体论的模型
基因本体论是由研究人员提出的用于描述基因及其产物特性的术语,而蛋白质就是后者的代表物质。该术语能够从细胞组分、分子功能和生物学过程三个角度描述蛋白质。由于具有相似功能的两个蛋白质更可能通过彼此发生相互作用,来承担细胞体内的同一个生理功能,而基因本体论又提供了获取蛋白质功能信息的相应途径,所以,可以通过某些方法计算蛋白质对的基因本体论相似性,从而评估它们的功能相似性,进而预测蛋白质相互作用。虽然有研究表明基于基因本体论的特征比基于序列的频谱计数特征具有更好的性能。但是,此类模型多数没有考虑细胞组分信息以及基因本体论类别层次结构中不相等的深度对于蛋白质相互作用预测带来的影响。
基于深度学习的模型
深度学习是近年来兴起的一项新技术,当被应用于预测蛋白质相互作用时,深度学习技术强大的学习能力能够更准确、更自动化地学习蛋白质的特征,从而生成更精确的特征向量,基于深度学习的模型不仅能够在一定程度上提升对于相互作用的预测能力,而且可以进一步减少人力的消耗。该类模型和基于序列的模型原理类似,都是从蛋白质中提取某些和相互作用相关的信息作为特征向量,然后利用这些特征向量结合现有的分类器模型评估两个蛋白质之间存在相互作用的概率。与上述几种类型的预测模型相比,基于深度学习的模型能够发挥深度学习技术的优势,挖掘潜在的有价值的蛋白质特征信息,使得模型的预测性能更好。
大规模预测模型
目前,已经鉴定出的蛋白质相互作用的数据还不到整个相互作用组的20%。随着高通量技术的发展,蛋白质相互作用数据的大小和复杂性也大大增加。大规模预测模型通常采用分布式的方法,并行预测蛋白质相互作用,以提高模型的效率,但由于各类模型所采用技术的局限性,并不是所有大规模预测模型都能保证预测性能优秀且预测效率高效。
实验方案和性能评估
实验方案和性能评估
实验方案
为了实现准确预测蛋白质相互作用,现有的计算模型通常遵循有监督的学习框架,准备相互作用和非相互作用的蛋白质对数据。其中,相互作用数据是阳性样品,非相互作用是阴性样品。前者可以从数据库中明确提取,后者可利用随机生成策略、细胞定位策略和Negatome 2.0获取。
一旦获得了实验数据,下一步就是选择合适的方案进行性能评估。通常,实验数据可分为训练集和测试集,前者用于训练模型,后者用于验证模型性能。常被用来划分训练集和测试集的方案包括三种:随机子抽样验证、K折交叉验证、留一法交叉验证。
性能评估
要定量评估计算模型预测蛋白质相互作用的性能,可以使用四种评估指标:Matthew相关系数(MCC)、F1得分、AUC和PR-AUC。其中,MCC是一种平衡度量指标,不仅可以指示预测结果与真实结果之间的相关系数,还可以处理数据集不平衡的情况;F1得分是为了平衡查全率和查准率而被提出的,是它们的调和平均数;AUC是ROC曲线与坐标轴所围面积的值,而ROC曲线是以FPR为横轴,TPR为纵轴所做;PR-AUC是 PR曲线与坐标轴所围面积的值,而PR曲线是以查全率为横轴,查准率为纵轴所做。
在线预测工具
在线预测工具
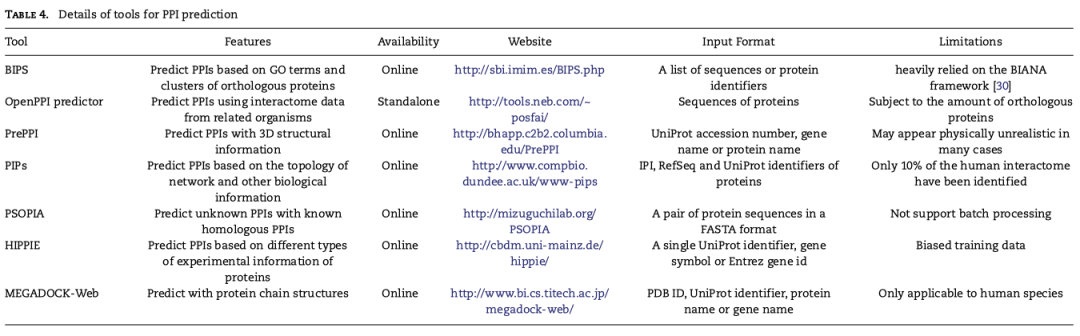
在线预测工具包括:BIPS、OpenPPI_predictor、PrePPI、PIP、PSOPIA、HIPPIE和MEGADOCK-Web。在这些工具中,它们能够通过蛋白质相关数据库以及生物信息对蛋白质相互作用进行预测。除了OpenPPI_predictor需要下载外,其他工具都直接提供了Web页面。
未来工作
未来工作
未来的挑战和工作可以从以下五方面展开:
(1)目前绝大多数预测模型都是遵循监督学习的范式提出的,训练集数据的质量是决定蛋白质相互作用预测准确性的关键问题。因此我们有必要在实验过程中对相互作用数据进行预处理,以确保数据的质量,从而更加准确地对预测模型性能进行评估。同时,除了相互作用数据外,我们还可以考虑蛋白质其他的生物学信息,来抵消对相互作用数据高度依赖导致的负面影响。因此,如何有效地整合多种生物信息资源以进行蛋白质相互作用预测仍然是未来需要解决的主要挑战之一。
(2)蛋白质相互作用在不同的细胞中和同一个细胞的不同周期中都是不同的,未来研究应该集中在预测动态相互作用上面。随着热邻近共聚技术(Thermal Proximity Coaggregation,TPCA)和下一代测序技术如RNA-seq的发展,我们可以利用TPCA特征和RNA-seq的时间序列数据设计新的动态预测模型。
(3)多数模型通常使用酵母或人类数据集评估蛋白质相互作用预测模型的性能,而存在共同进化的证据表明,相互作用的某些进化模式在不同物种之间是保守的。因此,未来的工作可集中在根据进化模式预测不同物种中的相互作用。
(4)由于高通量技术的发展,多组学数据可能为预测蛋白质相互作用提供额外的证据,但是相互作用与多组学数据之间的关系仍有待深入研究,如何有效地将多组学数据与机器学习技术结合起来是成功预测蛋白质相互作用的关键步骤。
(5)在基于网络预测蛋白质相互作用时,很少有模型会考虑局部网络结构和全局网络结构的互补性。此外,如何将蛋白质的生物学信息整合到相互作用网络中仍然是蛋白质相互作用预测需要解决的难题。随着属性图聚类算法的兴起,将局部和全局网络结构与属性图聚类算法结合可能是未来蛋白质相互作用预测的重点。
参考资料:
Lun Hu, Xiaojuan Wang, Yu-An Huang, Pengwei Hu, Zhu-Hong You, A survey on computational models for predicting protein–protein interactions, Briefings in Bioinformatics, 2021;, bbab036,
https://doi.org/10.1093/bib/bbab036
复杂科学最新论文
集智斑图顶刊论文速递栏目上线以来,持续收录来自Nature、Science等顶刊的最新论文,追踪复杂系统、网络科学、计算社会科学等领域的前沿进展。现在正式推出订阅功能,每周通过微信服务号「集智斑图」推送论文信息。扫描下方二维码即可一键订阅:

推荐阅读
点击“阅读原文”,追踪复杂科学顶刊论文
微信扫一扫,分享到朋友圈











