从蛋白质序列直接预测三维结构,AlphaFold 此前取得了突破性进展。而蛋白质与人类的语言有天然的相似性,蛋白质的氨基酸序列就像是语言的字母,语言模型有可能在整个演化过程中学习蛋白质序列的模式,是否可以用大语言模型预测蛋白质结构呢?在近日发表于 Science 的一项最新研究中,来自 meta AI 团队的研究者采用能够涌现出演化信息的大语言模型,开发了一个从序列到结构的预测器 ESMFold,对单序列蛋白的预测精度超过了 AlphaFold2,对有同源序列的蛋白的预测精度接近 AlphaFold2,且速度提升了一个数量级。该模型预测了6亿多条宏基因组的蛋白质,展示了天然蛋白质的广阔性和多样性。
关键词:大语言模型,蛋白质结构预测,共演化,宏基因组
刘贤 | 作者
梁金 | 编辑
Evolutionary-scale prediction of atomic-level protein structure with a language model
https://www.science.org/doi/10.1126/science.ade2574
在蛋白质科学中,序列决定结构,结构决定功能。从序列中直接预测结构,从而推断功能,是研究者50多年来的梦想。经过多年的探索,研究者发现可以用演化信息预测蛋白质的结构。近十年来,基于人工智能(AI)技术的发展、蛋白质序列数据和蛋白质结构数据的积累,用AI技术和演化信息对蛋白质结构的预测取得了突破,其中的典型代表是 Deep Mind 的 Alphafold2(AF2)。
对于大多数蛋白,AF2预测的结构基本可以和实验结构相当。但是,AF2对单序列蛋白的预测精度低,且预测所需的时间长。在本文中,来自 meta AI 团队的研究者采用能够涌现出演化信息的大语言模型,对单序列蛋白的预测精度超过了AF2,对有同源序列的蛋白的预测精度接近AF2,且速度提升了一个数量级。采用这个快速的模型,研究者预测了6亿多条宏基因组的蛋白质,展示了自然界蛋白质的广度和多样性。
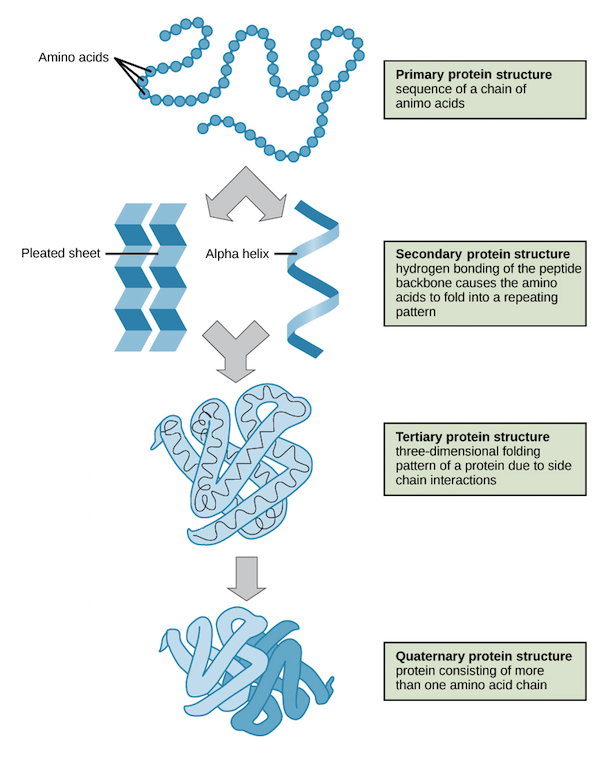
蛋白质由20种氨基酸通过肽键相连形成。稳定的蛋白质分子结构一般可划分为四个层次:
氨基酸之间通过共价键形成一条长链,为一级结构;
主链原子间的氢键形成特定的如β折叠、a螺旋等模式,为二级结构;
二级结构依靠残基的二硫键、疏水作用、范德华力以及离子键等非共价作用自发堆积在一起,为三级结构;
不同结构域之间依靠蛋白之间的相互作用形成四级结构(图1)。
蛋白质结构可以通过 X-晶体衍射实验、核磁共振(NMR)和冷冻电镜等实验技术鉴定。实验方法费钱、费时又费力,结构获取的速度远远跟不上蛋白质序列测定速度。因此,研究者一直试图直接通过计算的方法预测蛋白质的结构,即输入蛋白质的序列,输出蛋白质中每个原子的空间坐标。
图1. 蛋白质分子结构的四个层次 | 图片来源:https://www.khanacademy.org/science/biology/macromolecules/proteins-and-amino-acids/a/orders-of-protein-structure
早在20世纪70年代,Anfinsen 就提出“正常生理环境中的天然蛋白质三维构象处于整个系统中吉布斯自由能最低的状态;也就是说,蛋白质氨基酸序列包含了形成其热力学上稳定的天然构象所必需的全部信息”。因此,从理论上说,可以通过计算找到自由能最低的构象。但是,寻找自由能最低的构象所需的计算量过大,没有操作的可行性。
一种经典的蛋白质结构的预测方法为有模板建模,给定序列后,将序列分成多段,在已测定的结构数据库中搜索这些片段的结构,将这些结构组装在一起,即得到序列整体的三维结构。片段的结构是从数据库中搜索得到,有一定的准确性,片段的组装该如何完成呢?
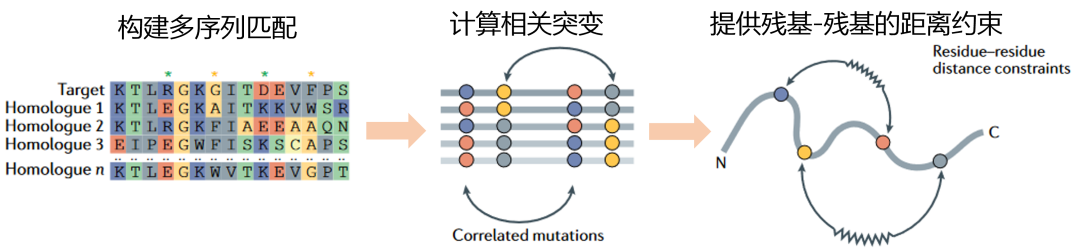
研究者发现,在演化中,如果两个氨基酸共演化(同时发生突变,图2),那么这两个氨基酸在空间上距离近。测序技术的发展积累了很多物种的蛋白质序列,可以进行搜库实现多序列匹配(图2)。组装的过程可以采用演化的信息对氨基酸-氨基酸的距离进行约束(图2)。共演化信息的运用,显著提高了结构预测的准确性。
图2. 共演化信息在结构预测中的运用。提供残基-残基的距离约束。| 改编自 Kuhlman, Brian, Philip Bradley. 《Advances in Protein Structure Prediction and Design》. Nature Reviews Molecular Cell Biology 20, 期 11 (2019年11月): 681–97. https://doi.org/10.1038/s41580-019-0163-x.
在结构预测的研究过程中,积累了近20万套的结构数据(PDB数据库)和上亿条蛋白质序列。过去十多年,深度学习技术快速发展,正好可以用来解决拥有大量数据的蛋白质结构预测问题。在CASP12(2016年)上,许锦波教授首次成功把残差网络(ResNet)应用于蛋白质残基接触图的预测中,大幅提升了残基接触图预测(contact map)的精度,为提升从头结构预测的精度奠定了基础。在CASP13上(2018年),DeepMind 团队基于类似思想开发了AlphaFold,在比赛中夺得冠军。在2020年举办的CASP14中,DeepMind开发的AlphaFold2震惊了世界,该模型采用注意力机制(Transformer),对竞赛的目标蛋白的预测精度GDT_TS(Global Distance Test – Total Score)中位数超过了90(图 1‑3),意味着对其中很多蛋白所预测的结构与实验结构非常接近,均方根差(Root Mean Squared Deviations,RMSD)在1-2埃以内。尽管AF2取得了巨大的成功,但由于AF2需要搜库构建 MSA(multiple sequences alignment),速度慢。发展新的速度更快的模型,非常有必要。
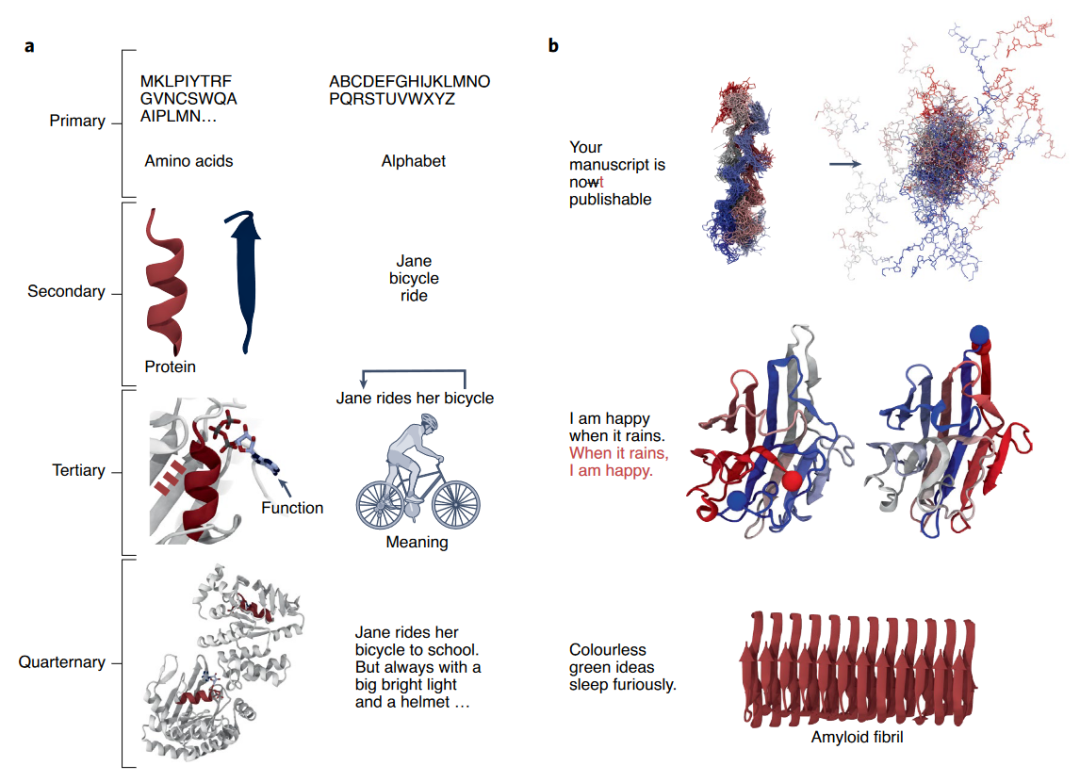
蛋白质与人类的语言有天然的相似性(图3)。语言的字母可以对应到蛋白质的氨基酸序列,语言的字母组成单词可以对应到氨基酸构成二级结构,语言中单词组成有意义的句子可以对应到二级结构组成蛋白质的三级结构,有意义的句子组成复杂的文本可以对应到多个蛋白组成四级结构(图3-a)。另外,语言中字母错配导致语义改变对应于氨基酸的改变导致蛋白质不能够正确折叠,字母片段和氨基酸片段顺序改变可以不改变语义和功能,可以生成合乎语法但无意义的句子对应于可以生成折叠成四级结构但没有功能的蛋白(图3-b)。
图3. 蛋白质和人类语言的相似性。| 引自Ferruz, Noelia, Birte Höcker. Controllable Protein Design with Language Models. Nature Machine Intelligence 4, 期 6 (2022年6月): 521–32. https://doi.org/10.1038/s42256-022-00499-z.
既然蛋白质与语言具有天然的相似性,通过大语言模型应该可以学习到蛋白质的结构信息,从而进行结构预测。来自 meta AI 团队的研究者对此进行了研究,训练了涌现出结构信息的大语言模型ESM,并开发了一个从序列到结构的预测器ESMFold,对单序列蛋白的预测精度超过了AF2,对有同源序列的蛋白的预测精度接近AF2,且速度提升了一个数量级。采用这个快速的模型,研究者预测了6亿多条宏基因组的蛋白质,展示了自然界蛋白质的广度和多样性。
1. 大语言模型涌现出原子级别的蛋白质结构
在本研究中,随机将序列中15%的氨基酸以掩码表示,模型的任务是根据掩码周围的氨基酸预测出掩码表示的氨基酸。模型可以学习到氨基酸之间的依赖关系,学习在演化过程形成的模式。模型训练采用的序列来自UniRef数据库。训练中,将1.38亿条序列聚类为0.43亿类别,等概率地从这0.43亿类别中取序列进行训练,因此,在整个训练过程中,模型共“看”了0.65亿条独特的序列。
研究者训练了一系列参数量从800万到150亿的多个蛋白质语言模型。随着模型增大,预测氨基酸的准确度提高。准确度可以通过困惑度(perplexity)来度量,直观来说,困惑度描述了对于序列中的每个位置,模型平均从多少个氨基酸中选择一个。困惑度从1到20,1表示完全准确的预测,20表示完全随机的预测(共有20种氨基酸)。对于同一个模型,随着模型训练步数的增加,困惑度减小;对于不同参数的模型,在相同的训练步数下,模型参数量越大,困惑度越小。800万参数模型的困惑度为10.45,而150亿参数的模型达到了6.37,表明在理解蛋白质序列上,模型越大,理解越准确。
图4. 当语言模型增大到150亿参数时,结构信息在模型中涌现
随着模型的增大,从中涌现出了蛋白质的结构特征。ESM2仅仅是基于序列训练,因此,任何出现的结构信息都是序列中模式的一种表示。已有的研究表明,Transformer 的蛋白质语言模型可以发展出与残基-残基接触图(残基-残基接触图表示蛋白质中两两氨基酸之间是否接触)相关的注意力模式。通过一个简单的线性投影,就可以从模型输出的注意力模式(attention pattern)中提取出残基-残基接触图(图4-A)。随着模型的增大,残基图的预测越准确(图4-B)。在演化上同源蛋白越多的蛋白,随着模型的增大,准确性饱和越快;而演化上同源蛋白越少的蛋白,准确性饱和越慢(图4-C)。
通过看一个一个的蛋白,研究者发现,接触图预测的准确性随着模型的增大非线性地提高。可视化从一个尺度到下一个更大尺度的模型的长程接触图预测的准确度,发现准确度是跃升的(图4-D)。接触图的预测准确度与困惑度是关联在一起的,接触图预测准确度的巨大变化也意味着困惑度的巨大变化。这种关联也预示着语言模型的目标是直接与结构信息在注意力图中的呈现(materialization)直接相关。
为了从大语言模型的蛋白质表示中得到原子级别的结构信息,研究人员采用了等变transformer(equivariant transformer)搭建了一个预测的模型。
在AF2中,在进行结构预测前,需要从数据库中搜索相似的序列、进行多重序列比对,这样的操作大概需要10分钟。本研究的大语言模型将演化模式内化在了模型中,因此不再需要外部的数据库、多重序列比对和模板,从而显著提高了结构预测的效率。
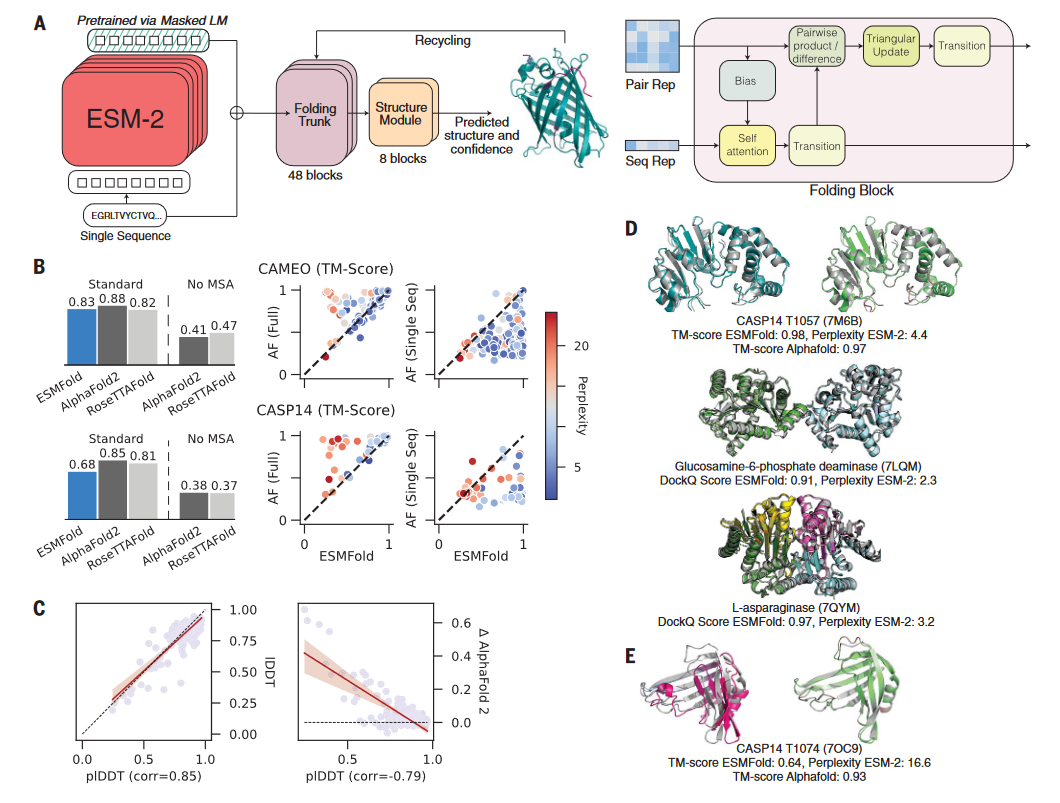
通过在ESM-2的基础上训练一个折叠的模型,开发了一个端对端的单序列结构预测器ESMFold。在预测的时候,蛋白质序列输入到ESM-2中,输出的隐向量输入到预测模块中。预测模块的第一部分是一序列的折叠块,每个折叠块在更新序列表示和两两表示。该部分的输出输入到等变 transformer 的结构模块中,通过3步的重复(recycling),最后产生结构和可信度。该预测模型与AF2的预测模型有相似之处,但更简洁(图5-A)。
与AF2相比,ESMFold的速度更快。在单个NVIDIA V100的GPU上,ESMFold预测一个长度为384的蛋白的结构只需14.2秒,比AF2快了6倍。对于更短的蛋白,ESMFold的速度可以比AF2快60倍左右。ESMFold快的主要原因是其不需要进行搜库构建MSA(大概需要10分钟)。
与AF2相比,ESMFold的平均准确性低(图5-B)。在CAMEO和CASP14的数据集上,AF2的TM-score分别为0.88和0.85,而ESMFold的TM-score分别为0.83和0.68。
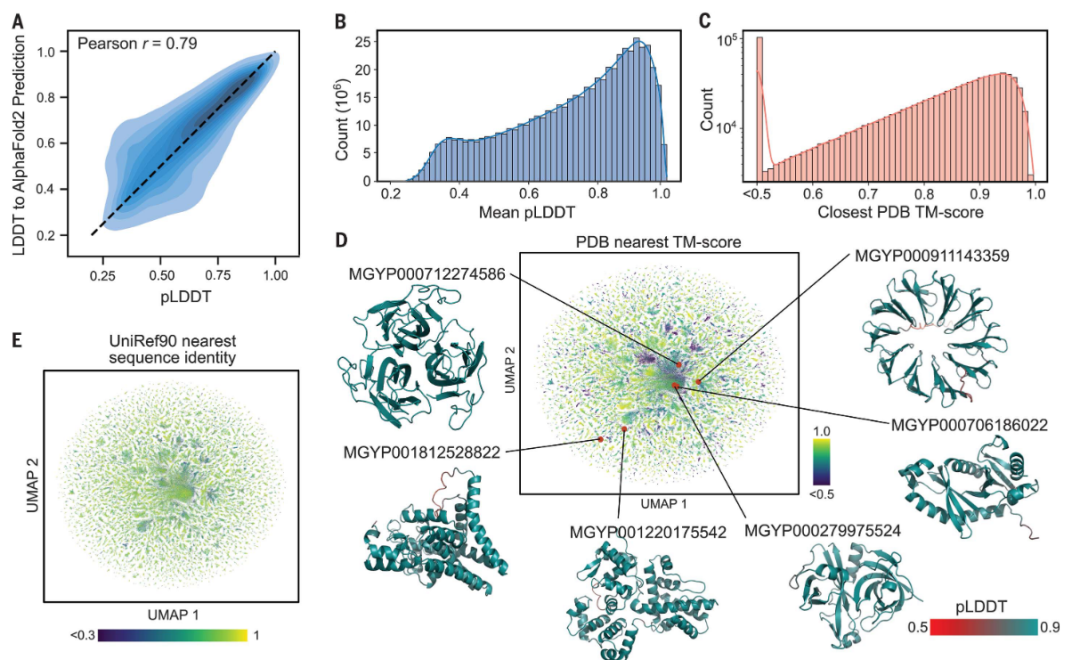
高精度且快速的结构预测能力为宏基因组蛋白质结构的刻画提供了机会。该研究预测了MGnify90数据库中长度20~1024的6.17亿多条序列的结构,占到所有序列的99%。总结来说,研究预测大约3.65亿条是中等可信度的(good confidence),占到59%,有2.25亿条是高可信度的(high confidence),占到36%。
ESMFold 的可信度分数是一个与实验结果和 AF2 结果相符的很好的指标。研究者在宏基因组的蛋白上评估了ESMFold 和 AF2。在大约4000条宏基因组的蛋白质上,ESMFold 和 AlphaFold2 的 pLDDT 具有高相关性(Pearson r = 0.79)。结合在 CAMEO 数据上的结果,当可信度非常高(pLDDT > 0.9)时,ESMFold预测能够达到实验的准确度。这些结果表明,在所有预测的6.17亿条蛋白质中,1.13亿条满足超高可信度的标准(pLDDT > 0.9)。
ESMFold 可以高效地刻画远离已有知识的蛋白质空间。很多高可信度的宏基因组蛋白的结构不在已有的数据库中。在随机的一百万条高可信度蛋白结构中,76.8% 的蛋白与 UniRef90 中的任一蛋白的序列同一性低于 90%,表明它们与已有的 UniRef90 序列非常不同;3.8% 的蛋白在 UniRef90 中一条相似的蛋白都没有找到。用 Foldseek 比较预测的结构与PDB数据库中的结构,在 TM-score 的卡值为0.7和0.5时,分别有 25.4% 的蛋白和 12.6% 的蛋白没有找到相似的蛋白结构;在 TM-score 的卡值为0.5、序列一致性的卡值30%时,有2.6%蛋白没有找到相似的蛋白结构。
大规模的结构刻画使缺失序列相似性的情况下鉴定结构相似性成为可能。很多高可信度的结构对应的序列与UniRef90 中的序列相似性低,却在PDB数据库中具有相似的结构。这种遥远的同源超出了通过序列一致性可以检测的范围。比如,MGnify 的序列 MGYP000936678158 在 UniREF90 中没有匹配的序列,通过 jackhmer 也没有搜索到相似的序列,但它却有在多个核酶中保守的结构。类似地,MGYP004000959047 在 UniREF90 没有匹配的序列,和 jackhmer 的搜库结果也不相似,却与脂结合结构域具有相似的结构。通过结构检测远距离相似性的能力可以为功能研究提供新的见解,而这些见解从序列中是得不到的。
所有预测的结构可以通过ESM Metagenomic Atlas (https://esmatlas.com) 获取。通过API可以批量下载结构,可以通过结构和序列进行在线搜索。
AI+Science 是近年兴起的将人工智能和科学相结合的一种趋势。一方面是 AI for Science,机器学习和其他 AI 技术可以用来解决科学研究中的问题,从预测天气和蛋白质结构,到模拟星系碰撞、设计优化核聚变反应堆,甚至像科学家一样进行科学发现,被称为科学发现的“第五范式”。另一方面是 Science for AI,科学尤其是物理学中的规律和思想启发机器学习理论,为人工智能的发展提供全新的视角和方法。
集智俱乐部联合斯坦福大学计算机科学系博士后研究员吴泰霖(Jure Leskovec 教授指导)、哈佛量子计划研究员扈鸿业、麻省理工学院物理系博士生刘子鸣(Max Tegmark 教授指导),共同发起以“AI+Science”为主题的读书会,探讨该领域的重要问题,共学共研相关文献。读书会从2023年3月26日开始,每周日早上 9:00-11:00 线上举行,持续时间预计10周。欢迎对探索这个激动人心的前沿领域有兴趣的朋友报名参与。
2022年11月30日,一个现象级应用程序诞生于互联网,这就是OpenAI开发的ChatGPT。从问答到写程序,从提取摘要到论文写作,ChatGPT展现出了多样化的通用智能。于是,微软、谷歌、百度、阿里、讯飞,互联网大佬们纷纷摩拳擦掌准备入场……但是,请先冷静一下…… 现在 all in 大语言模型是否真的合适?要知道,ChatGPT的背后其实就是深度学习+大数据+大模型,而这些要素早在5年前的AlphaGo时期就已经开始火热了。5年前没有抓住机遇,现在又凭什么可以搭上大语言模型这趟列车呢?
集智俱乐部特别组织“后 ChatGPT”读书会,由北师大教授、集智俱乐部创始人张江老师联合肖达、李嫣然、崔鹏、侯月源、钟翰廷、卢燚等多位老师共同发起,旨在系统性地梳理ChatGPT技术,并发现其弱点与短板。本系列读书会线上进行,2023年3月3日开始,每周五晚,欢迎报名交流。

https://pattern.swarma.org/study_group/23?from=wechat