巴拉巴西新作:AI-Bind 助力蛋白质-配体结合预测

关键词:药物发现,机器学习,网络拓扑,蛋白质-配体二分网络


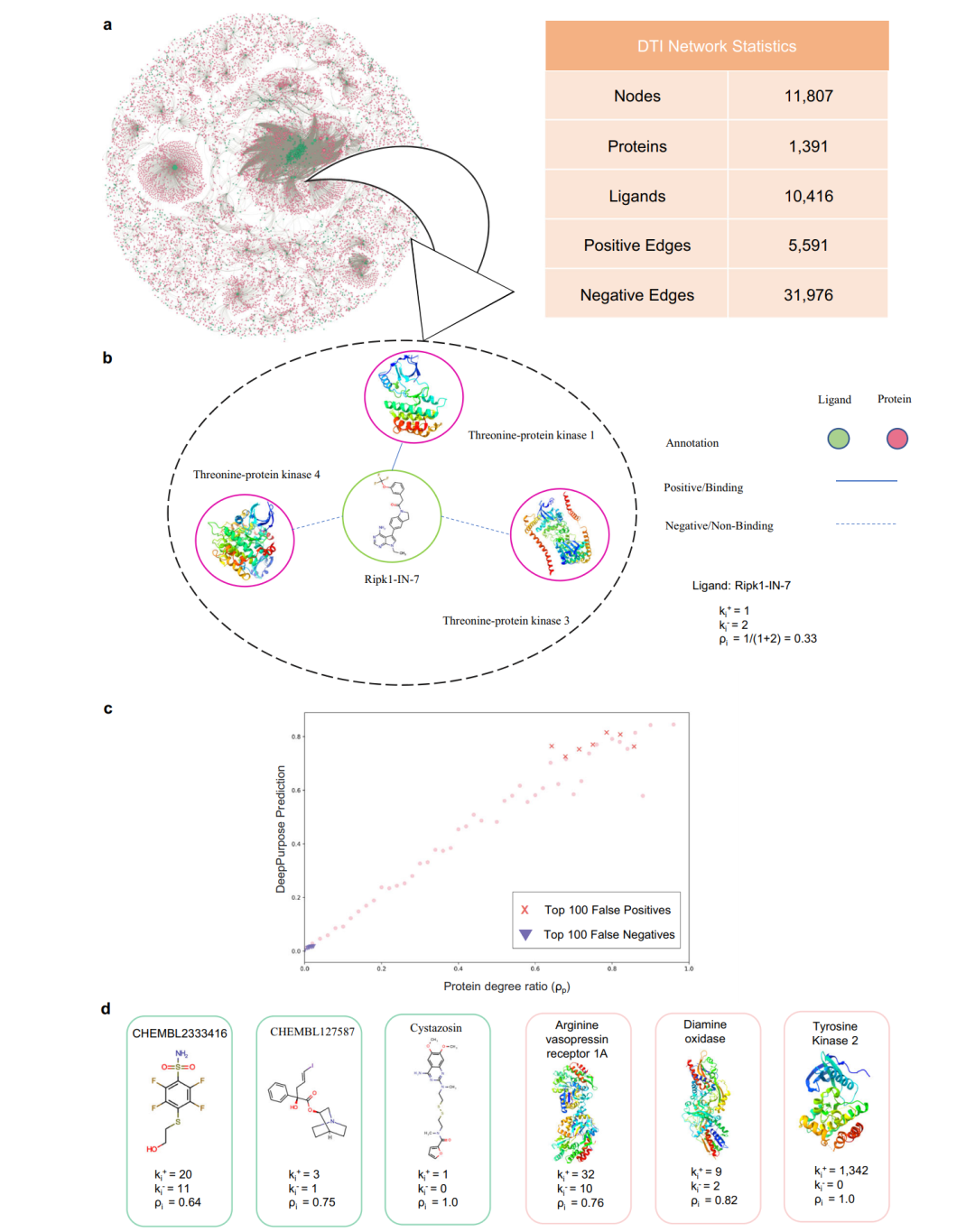
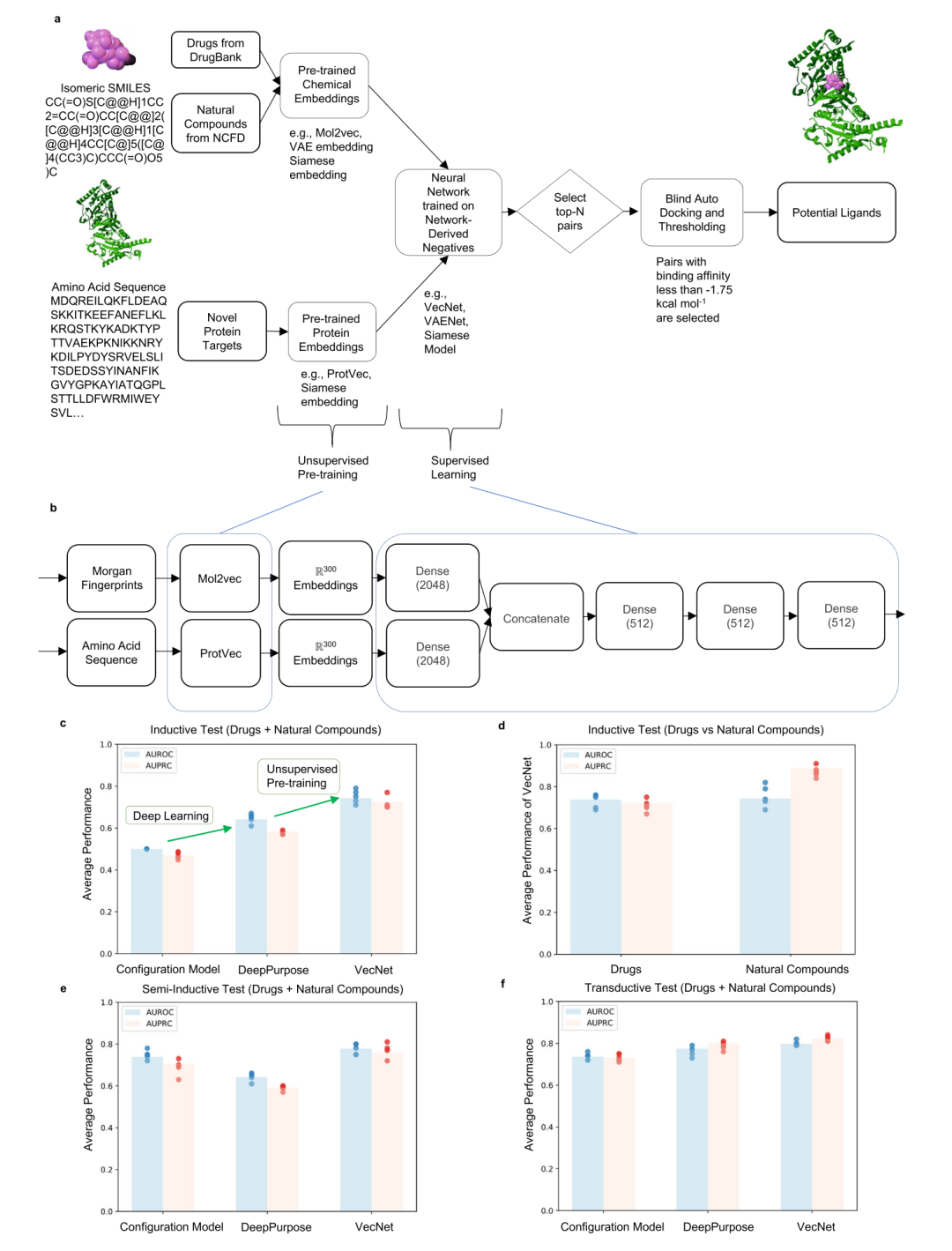
论文题目:Improving the generalizability of protein-ligand binding predictions with AI-Bind 论文来源:Nature Communications 论文链接:https://www.nature.com/articles/s41467-023-37572-z

AI+Science 读书会启动

推荐阅读
微信扫一扫,分享到朋友圈











