系统生物学中的建模:因果理解先于预测?

导语
技术的进展为我们提供了大规模的生物数据,但要如何从中获得对生物学机制的理解?一方面我们可以使用来自科学文献的先验知识,但这种知识驱动方法往往基于特定的情境,不适合进行因果关系预测;另一方面,纯数据驱动的推断方法专注于创建特定情境下的预测模型,却很难进行有意义的生物学解释。
2021年6月,发表于Cell 旗下Patterns 杂志的论文“蛋白质组中的因果相互作用:分子数据遇见通路知识”,介绍了一款名为 CausalPath 的因果推断工具,能够结合新的测量结果与先验知识,推断蛋白质组数据中因果信号的相互作用。这种方法模仿了生物学家用先验知识解释新的实验数据的传统方法,但可以在数十万反应的规模上进行。以下是 Patterns 杂志对这项工作的评论。
研究领域:系统生物学,计算生物学,因果推断

Szilvia Barsi, Bence Szalai | 作者
王百臻 | 译者
梁金 | 审校
邓一雪 | 编辑

论文题目:
Causal interactions from proteomic profiles: Molecular data meet pathway knowledge
论文链接:
https://www.cell.com/action/showPdf?pii=S2666-3899%2821%2900083-0
1. 大规模生物数据集该如何解释?
1. 大规模生物数据集该如何解释?
高通量技术的最新进展允许我们获取不同模式的大规模生物数据集,如转录组学、蛋白质组学或代谢组学数据集,甚至在单细胞水平上也是如此。虽然这些数据集为我们提供了独一无二的视角,来了解生物体内健康与疾病表现型背后的分子机制,但由于多种不同因素的相互作用,获取对它们的正确解释却变得异常复杂。
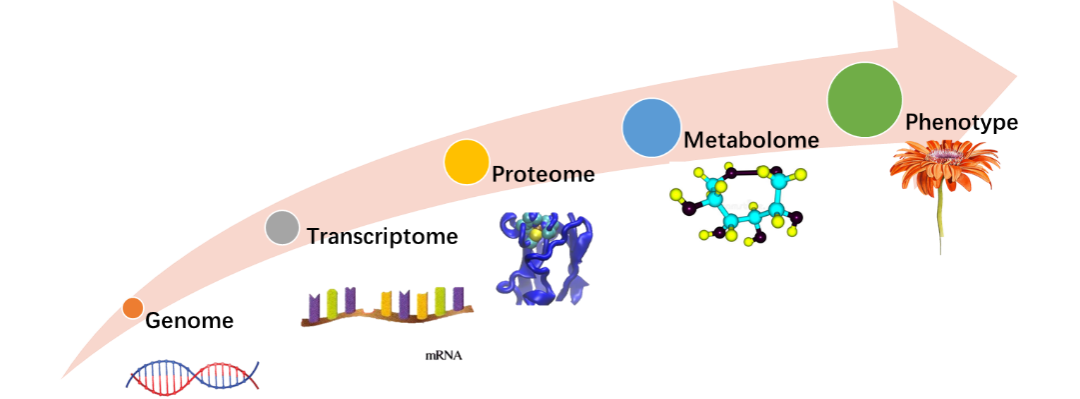
图1. 基因组、转录组、蛋白质组、代谢组、表现型
首先,在大多数情况下,标准的分析方法只会返回冗长的差异表达基因列表,或表型相关基因列表,乃至蛋白质列表,这阻碍了我们深入理解观察到的表型的背后机制。此外,实验数据的高维数(例如转录组学中数据维数可能超过20,000维),使区分简单的相关性和复杂的因果性变得更为复杂——而对于理解其中机制并将其用于干预治疗中,对因果关系的理解恐怕必不可少。
2. 建立系统生物学模型
2. 建立系统生物学模型
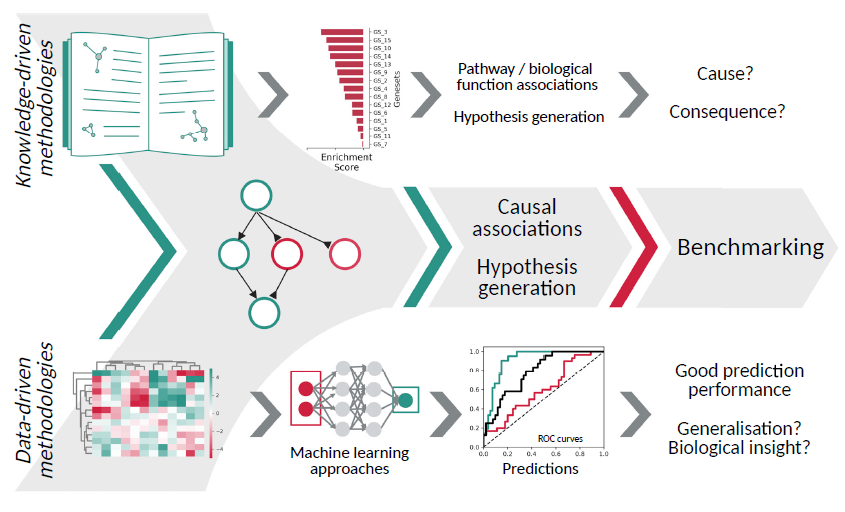
为了克服这些限制,人们建立了不同的系统生物学模型及分析技术。通常,这些方法大体可以归为两类:知识驱动的方法与数据驱动的方法。
在大多数情况下,知识驱动的方法使用经过筛选的基因集列表,以生成环环相连的生物过程或路径,并使用统计方法来发现生物数据集中,这些基因集里可能存在的过度表达或富集现象。相比于简单的差异表达基因列表,知识驱动的方法往往可以提供更多的生物学见解,因此更适合于假设生成。然而,在大多数情况下,研究使用的基因集往往过于笼统,以致我们无法从数据中识别真正的因果信息。
另一方面,包括机器学习模型在内的数据驱动方法专注于预测性能。从以下几个方面来看,系统生物学模型的预测性能很重要:首先,从药物发现到患者分层等不同生物学领域,预测模型都扮演着很重要的角色。此外,有人可能会争辩说:如果某些生物表型可以从组学数据中预测,这意味着预测模型确定了潜在的生物机制。然而,不幸的是,这些后来提出的主张被高估了:机器学习模型可以通过机器学习方法,分析数据集中的一些技术偏差和混杂因素,这可以有效地提高预测性能,但同时也会阻碍我们进行生物学意义上的解读以及理论推广。此外,相当一部分表现最好的机器学习模型属于“黑盒”模型,这意味着从中推导出准确的预测机制是一件很困难的事,这也阻止了我们进行有意义的生物学解释。
3. 因果推断的新工具
3. 因果推断的新工具
最近,研究人员开发了几种新方法来弥合知识驱动方法与数据驱动方法之间的差异。这些“因果推断工具”将先验知识网络(如信号通路或基因调控网络)与基因组规模的基因表达或蛋白质组学测量连接起来,并使用统计工具来识别情境化的、特定于样本的信号网络变化趋势,从而解释观测数据的因果效应。相比于不同基准的经典知识驱动方法,这些方法已被证明能够更好地估计通路活性变化。
巴布尔等人[Babur et al. (2021) ]为这个后来的工具集添加了一种有趣的新方法,能够用来区分简单的相关性和较为复杂的因果性。CausalPath 方法使用来自路径共用数据库(Pathway Commons database)的激酶/磷酸酶底物*和转录因子所调节的基因关系,来创建图形模式。这些图形模式是类似下面这样的因果关联:当P1位点磷酸化时,激酶A是活跃的;活性激酶A在P2位点上磷酸化蛋白质B。这些图形模式与如下测量结果相匹配:激酶A在P1位点磷酸化,而蛋白质B在P2位点磷酸化;从而使我们可以做出因果推测,识别信号的潜在因果方式:给定数据集中激酶A磷酸化蛋白质B。
*译注:激酶是磷酸化酶,可利用能量分子(如ATP)将磷酸基团加到对应底物分子上;磷酸酶的作用与激酶相反,能够将对应底物去磷酸化。
CausalPath 方法还使用基于数据标签排列的方法检验结果的统计显著性。在他们的论文中,作者测试了不同癌症相关数据集的方法,成功地从蛋白质组学数据中识别了不同配体和药物的作用机制。
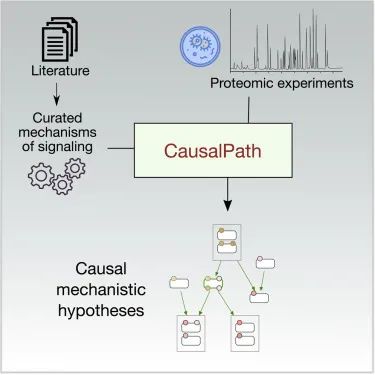
图3. CausalPath 方法结合来自科学文献中已有的关于生物通路的详细知识与最新获得的蛋白质组学和其他分子测量数据,生成机制模型来解释观测到的变化如何彼此关联。
巴布尔等人的研究结果还强调了将正确类型的先验知识与相对应的组学模式结合使用的重要性。
当他们将基因调控网络与蛋白质组学数据结合使用时,推断出的因果网络在统计学意义上并不显著,而将相同的先验知识网络与基因表达数据结合使用,会导致更加显著的因果关联。这些结果还突出了目前系统生物学建模的一个普遍存在的问题:鉴于转录组学数据集的丰度更高(例如,与磷酸蛋白质组学相比),基因表达数据更常用于建模研究。然而,在大多数情况下,使用的先验知识网络是在蛋白质活动的水平上定义的。由于基因表达与蛋白质丰度及活性之间的关联可能不大,因此将基因表达数据与通路网络结合使用,可能会对结果给出错误解释。
这些思考以及巴布尔等人的研究结果,共同表明了一个至关重要的问题——需要使用匹配的先验知识网络和数据,例如:基因调控网络与转录组学,信号网络与蛋白质组学。正确整合不同类型的先验知识网络和数据类型,也有望能够识别多组学数据集中的因果关系。
4. 基准测试
4. 基准测试
虽然目前因果推理方面最重要的工具是生物假设生成,但为了对不同方法进行基准测试以选择性能最佳的方法,评估预测性能或因果推理工具也是至关重要的。在巴布尔等人的论文中,他们将自己的方法与几种现存的方法进行了比较,这是朝着这个方向努力所迈出良好的第一步。
当然,随着越来越多的相关工具被开发出来,执行无偏见、独立的基准测试至关重要。基准测试的一个重要瓶颈是因果关系已知的高质量数据集。为此,扰动数据集(变化的一般原因已由使用的扰动所给出,例如药物、基因操作等)看起来最为合适,但扰动的偏离目标效应会使方法的评估复杂化。尽管如此,诸如DREAM挑战(Dialogue on Reverse Engineering Assessment and Methods Challenges)等大型基准测试项目,可以促进未来因果推理系统生物学工具的开发与评估。
参考资料
(参考文献可上下滑动查看)
因果科学读书会第三季启动
由智源社区、集智俱乐部联合举办的因果科学与Causal AI读书会第三季,将主要面向两类人群:如果你从事计算机相关方向研究,希望为不同领域引入新的计算方法,通过大数据、新算法得到新成果,可以通过读书会各个领域的核心因果问题介绍和论文推荐快速入手;如果你从事其他理工科或人文社科领域研究,也可以通过所属领域的因果研究综述介绍和研讨已有工作的示例代码,在自己的研究中快速开始尝试部署结合因果的算法。读书自2021年10月24日开始,每周日上午 10:00-12:00举办,持续时间预计 2-3 个月。

详情请见:
因果+X:解决多学科领域的因果问题 | 因果科学读书会第三季启动
推荐阅读
-
Donald Rubin的因果推断学术贡献:超出统计学范畴的划时代影响 -
Physics Reports计算网络生物学长文综述:数据、模型和应用 -
丁鹏:多角度回顾因果推断的模型方法 -
PNAS:大脑如何整合多感官模态信息,进行因果推断? -
加入集智,一起复杂!
点击“阅读原文”,报名读书会
微信扫一扫,分享到朋友圈











