Nat. Methods | 利用深度学习进行基于生物物理学和数据驱动的分子机制建模

导语
本文介绍由美国马萨诸塞州波士顿哈佛医学院系统生物学系系统药理学实验室的Mohammed AlQuraishi等人发表于Nature Methods 的研究成果:研究人员报道了可微程序与分子和细胞生物学结合产生的新兴门类:“可微生物学”。本文作者介绍了可微生物学的一些概念并作了两个案例说明,展示了如何将可微生物学应用于整合跨生物实验中产生的多模态数据,解决这一存在已久的问题将促进生物物理和功能基因组学等领域的发展。作者讨论了结合生物和化学知识的ML模型如何克服稀疏的、不完整的、有噪声的实验数据造成的限制。最后,作者总结了它面临的挑战以及它可能扩展的新领域,可微编程仍有很多可发挥的空间,它将继续影响科技的发展。
 DrugAI | 来源
DrugAI | 来源
1. 简介
1. 简介
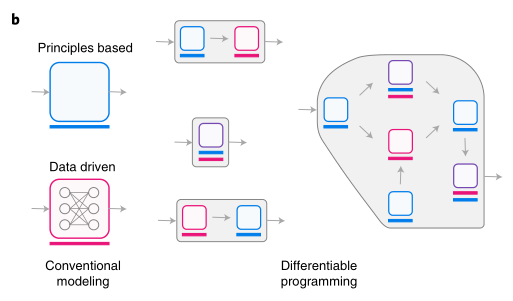
得益于神经网络和深度学习的快速发展,机器学习(ML)及其在人工智能(AI)中的应用在过去十年中经历了巨大的变化。深度学习演变出了一类可学习模型:可微程序,可微编程的出现是基于基础科学和软件工程中的四个要素:更好的模式识别器、定制建模的兴起、联合优化和稳定的可微编程框架(例如TensorFlow、Pytorch和JAX),它在分子和细胞生物学、蛋白质结构测定和转化生物医学等领域产生了深远的影响。
可微程序可以将针对特定自然科学领域的数学方程与通用的、基于实验数据训练的机器学习组件相结合。将可微程序用于对分子和细胞生物学的研究,形成了新兴的“可微生物学”,在可微生物学中,对小型、特殊(例如一次实验试验)抑或是一般、复杂(例如蛋白质折叠)的问题都可以有效地建模,并且利用基础科学知识能够克服稀疏的、不完整的、有噪声的数据所带来的限制。端到端的可微程序的一个重要特点是,它们可以进行从输入到输出的联合优化。这使得将传统上需要单独处理的任务(如数据预处理)统一到模型中成为可能。预处理需要对原始数据进行转换,再根据经验提取重要的信息。这种类型的“特征工程”通常是不透明的,缺乏记录文档,并且难以扩展。而可微程序不强调特征工程,而是透明地将现象建模到学习系统中,包括引入抽象概念,如溶液中分子的平移或旋转不变性、多肽骨架的化学性质等。为了实现这一点,可微程序使用了从物理学和化学的传统模型中得出的逻辑和方程。另外,不同于传统数学模型使用少量拟合变量(其中许多具有物理意义)进行参数化,可微程序的可学习组件(例如神经网络)可以有数百万甚至数十亿个变量。训练(优化)包括学习这些参数。
本文作者介绍了可微生物学的三个概念:生物模式、机械先验和数据先验,并作了两个案例说明:蛋白质结构预测、均匀化PPI数据,展示了如何将可微生物学应用于整合跨生物实验中产生的多模态数据,解决这一存在已久的问题将促进生物物理和功能基因组学等领域的发展。作者还总结了它面临的挑战以及它可能扩展的新领域,可微编程仍有很多可发挥的空间,它将继续影响科技的发展。
2. 结果
2. 结果
可微生物学的三个概念
在生物学背景下,可微编程有三大概念原语:生物模式、物理和机械或生物物理先验以及实验和数据采集先验。先验对模型进行了限制,并允许使用较小的数据集,最有用且可靠的先验是基于已被充分接受和理解的物理和化学系统特征的先验,例如多肽链中允许的键角范围。从过于复杂、难以理解而无法显式建模的数据映射中学习的模式识别器,可以与先验结合使用。
生物模式
模式识别器是最成熟的可微编程工具,目前它已应用于学习视网膜基底、识别并分割拥挤环境(如组织)中的细胞边界,以及从多重免疫荧光图像中预测新细胞的状态。模式识别器的复杂性通常由输入的结构决定,固定尺寸的二维(2D)网格表示的图像(例如由传统电子相机收集的图像)是最简单的输入,并且具有平移不变性,可以通过数据增强作为模型训练中的先验信息。‘图像’不需要局限于视觉模式。例如,编码残基协同进化的蛋白质内接触图谱已被用作卷积神经网络(CNN)的输入来预测蛋白质结构。将2D网格推广到更高的维度,例如,通过将3D空间离散成大小相等的立方体,,已经产生了可以对高分子量大分子进行操作的模式识别器,能够预测蛋白质功能和蛋白质-药物复合物的亲和力。
尺寸随输入数据而变化的可变尺寸网格(例如由不同长度的DNA序列组成的一维网格),是复杂性的再次提升。树和其他类型的图,可以表示交互网络和分子,不仅在长度上不同,而且在结构上也不同,可以使用图卷积网络(GCN)进行学习。GCN已被用于学习从分子到蛋白质结合亲和力的映射,并用于硅化学逆合成。综上所述,神经网络的关键优势在于它能够识别在小尺度和大尺度上发生的多向相互作用。
大多数当代ML应用程序关注复杂输入(如蛋白质结构)和简单输出(如结合亲和力)之间的关系,而可微编程允许更复杂的输入-输出映射。作者开发了一种可微递归几何网络(RGN),它直接从序列中学习蛋白质结构,将可变长度的蛋白质序列作为输入,并生成可变大小的原子坐标集作为输出。最近,由谷歌的姐妹公司DeepMind开发的AlphaFold2使用可微系统预测单域蛋白质结构,其准确度接近实验方法(图1)。从简单输入(一级序列)生成复杂输出(例如3D折叠蛋白质)的能力证明了可微程序相对于传统ML方法的一个主要优势。后者的范围仅限于简单输出类型,最常见的是分类变量或实值标量。

机械先验
生物学中的ML研究越来越多地将有关结构、化学和进化的先验知识纳入可微程序(图2a)。先验信息的范围从基因或蛋白质的计数及其相互作用到基本的生物物理学,包括了空间本身的特征。例如,大分子内部和之间的相互作用具有平移和旋转不变性,这可以通过将CNN从固定网格(不保证旋转不变性)推广到 Lie groups来实现, Lie groups可以捕捉三维(或更高)维的旋转对称性。在模拟蛋白质-蛋白质相互作用(PPI)网络、蛋白质折叠和类似复杂的生物现象时,这些先验知识的结合使捕捉与距离相关的物理相互作用(例如,旋转不变的静电力)变得更容易。
在生物医学应用中,定制ML模型的一个重要价值是,它包含了大分子结构和化学性质的详细信息。作者开发了一个模型,用于预测参与信号转导的肽结合域(PBD)的配体。通过在PBD家族中共享能量势,作者引入了共享和重复使用结合表面的概念,为这种大分子相互作用创造了一种灵活的的“通用语”。能量势是学习的,而不是规定的(只假设了重用的概念),且它可以提高模型的准确性,特别是在数据贫乏的领域。这里的PBD-配体相互作用模型是完全可微的,但没有使用神经网络。
在对生物网络建模时,结合更专业的先验知识也是可行的。例如,转录、蛋白质组和磷酸蛋白质组时间序列数据的联合建模可以通过在磷酸化信号和转录调控之间施加时间间隔来增强。这种专业知识可以与特定信号通路(例如,MAPK激酶级联的结构)或转录因子结合基序的分子数据相结合(图2b).在这样一个假设模型中,可以推断出所有可能的蛋白质-蛋白质和蛋白质-DNA相互作用的矩阵,其中一些相互作用对已经预先确定(例如从文献或重点实验中获取)。
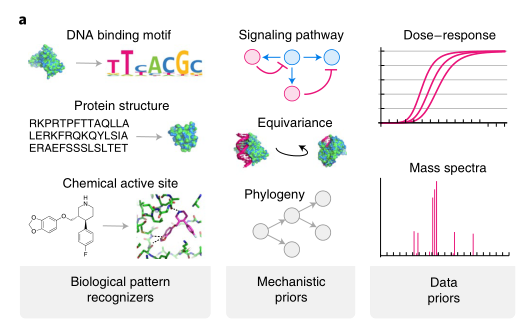
数据先验
生物学中的大多数建模涉及对不完整、有噪声、异构的数据的分析。为了将错误的影响降至最低并融合不同的数据类型,有必要结合体现数据生成过程的先验,数据规范化是另一个重要问题。
随机误差存在于所有真实世界的数据中,大多数分子测量也受到对系统误差知之甚少的影响。基于物理的误差建模在结构生物学和高分辨率光学显微镜学中很常见,在这些领域中,对测量过程和预期物理现象的范围有足够的了解,因此可以量化并建模许多的不确定性。虽然这种方法原则上可用于其他生物分析,但复杂的误差模型在生物医学研究中相对较少,通常是因为对潜在的物理过程没有充分的了解。在这种情况下,可以对误差进行简单的参数化。当目标生物物理量(如解离常数)来自间接测量时,这一点很重要。例如,蛋白质-蛋白质亲和力的实验表征涉及多种测量不同物理参数的分析方法(例如,开/关速率、平衡结合、结合所需的热量、活性抑制、底物之间的竞争等)简单和复杂的方程都可以用来描述实验观测值和潜在的生物物理参数之间的关系,这些方程可以合并到可微程序中。在优化过程中通过这些方程进行反向传播,使得较准确地估计未知参数成为可能,因为优化联合考虑了模型的所有方面。即使无法获得简单的分析公式,在神经网络中加入常微分方程(ODE)求解器的最新进展表明了将基本定律编码为可微程序中的微分方程的可行性。
两个实例
为了说明上述概念,作者介绍了可微生物学用来解究问题的两个案例。第一个案例说明了生物学先验知识是如何在定制ML模型中体现的;第二个案例则是讲PPI背景下的数据同质化。
蛋白质结构预测
蛋白质结构预测的目标是构建将蛋白质序列(离散符号的可变长度字符串)映射到蛋白质的三级结构(三维坐标的可变长度序列)的模型。最近的基于ML的方法利用了机械(生物物理)先验和模式识别。理论上,现成的模式识别器(如递归神经网络)可以完成这种映射,但在实践中,要达到较高的性能,则需要利用70年来解析蛋白质结构所获得的蛋白质几何特征(图3a)。例如,蛋白质骨架是共价键合的多肽链,其键长和角度几乎固定,但扭转角度依赖于序列。
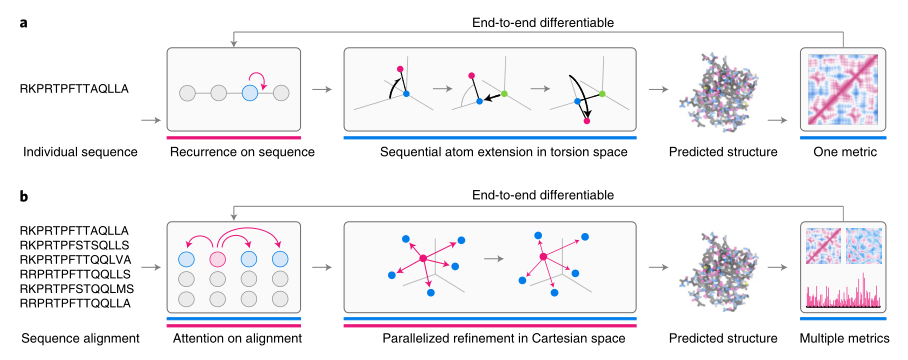
扭转角和三维坐标之间存在一对一映射。想要预测三维结构,从氨基酸序列预测扭转角,并优化模型参数以使预测的扭转角与已知扭转角之间的一致性最大化就可以了。固定键长和键角似乎只是简单的附加条件,但它对学习效率和准确性有重要影响,也有助于确保局部蛋白质几何结构的正确性。
然而,这种纯粹的局部方法仅通过预测扭转角的准确性进行训练和判断,效果不佳,因为微小的局部误差累积到一个完整的蛋白质上会产生较大的误差。更好的方法是将局部扭转角转换为三维蛋白质坐标,作为建模过程的一部分,使用能够最大化蛋白质数据库中预测坐标和已知坐标之间一致性的参数。
设计一个定制损失函数,还可以实现对蛋白质结构数据进行更复杂的处理,因为蛋白质结构数据经常会丢失(无序)侧链原子和序列延伸,这是因为非结构域是蛋白质功能的一部分。如果不考虑这些蛋白质,训练数据的数量最多会减少50%,而且可能会产生偏差。自定义损失函数可以自动忽略未解析的原子或残留物,因此,除了十几个蛋白质数据库中的大约100000个独特结构外,可以对其他所有结构进行训练(图3a)。该方法还适用于从单个蛋白质序列预测结构,而无需使用任何协同进化信息。
均匀化PPI数据
作者通过一个模型说明了可微程序在数据融合中的使用,该模型从不同时间使用不同方法收集的不同类型的实验数据中学习PPI亲和力(图4)。必须从不同类型的数据入手,每种数据都不完整,并且涉及不同的测量方法,第一类数据来源是定量生物物理方法直接测量,如表面等离子体共振(SPR)、等温滴定量热法(ITC)或肽阵列,这些是最定量但最不常见的数据类型。第二个数据来源是使用亲和纯化质谱或使用酵母双杂交分析(“高通量结合数据”)从细胞提取物中获取的。第三类数据涉及从基因或过表达研究推断的功能相互作用,这种数据很多,但最不直接。第四类数据包括单个PPI的高分辨率共复合结构,用这些来验证ML推断的生物物理假设。
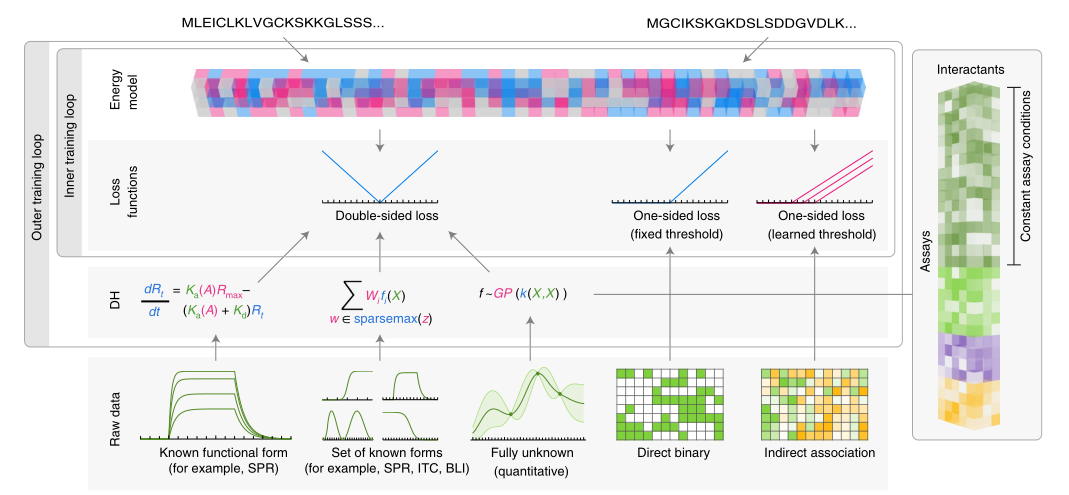
目标是利用全部可用数据创建一个模型,该模型使用具有两个组成部分的可微模型预测PPI的平衡结合常数(Ka):(1)将输入(蛋白质序列对)映射到预测Ka值的能量函数;(2)一组数据均质器,将原始实验数据映射为可与预测Ka值进行比较的形式。对整个模型进行联合优化,以使数据均匀化器中的能量函数的性能参数最大化(用于预测PPI亲和力)。
要考虑的最后一类数据涉及间接的相互作用,在现存的PPI数据库中涵盖了多种可能的检测,包括上位遗传相互作用、跨组织或细胞类型的基因或蛋白质共表达的模式和进化保护。虽然阳性分数不能证明蛋白质之间的物理相互作用,但与之正相关。(学习的)检测阈值取决于统计相关性的强度,因此不适用于单个测量。但是,如果相关性很高,模型可能能够提取有意义的信息。另一方面,如果相关性较低,则学习的阈值将设置为非常高的数字,在这种情况下,不会提取任何信息,并且模型不受影响。
上述方法的一个关键特征是,数据均匀器的参数与能量函数的参数一起学习。其中有一个问题,即用于均匀化数据的参数与通过能量函数学习的参数之间存在不必要的相互作用,可能导致退化解,在这种退化解中,由于所有均匀化参数都设置为零,性能会误导性地高。为避免此类问题,必须对数据均一器施加约束:参数必须为非零,并根据先验知识确定参数落在有意义的范围内。内部反向传播回路将能量势的参数拟合到训练集,而外部反向传播回路将数据均质器的参数拟合到验证集,并且通过第二个验证集评估整个过程。在这些情况下,需要两个验证集来避免过度拟合,正如在所有ML应用程序中一样,仔细选择培训、测试和验证数据集非常重要。
3. 总结
3. 总结
7、可微程序和其他形式的深度学习正在迅速发展,它们有望加速分子生物学等多领域的研究,而不仅仅是蛋白质结构预测。
总的来说,它并没有取代上一代基于机械或物理的模型,而是与这些模型融合,同时也处理了大量历史上难以解决的问题。
参考资料
AlQuraishi, M., Sorger, P.K. Differentiable biology: using deep learning for biophysics-based and data-driven modeling of molecular mechanisms. Nat Methods 18, 1169–1180 (2021).
https://doi.org/10.1038/s41592-021-01283-4
复杂科学最新论文
集智斑图顶刊论文速递栏目上线以来,持续收录来自Nature、Science等顶刊的最新论文,追踪复杂系统、网络科学、计算社会科学等领域的前沿进展。现在正式推出订阅功能,每周通过微信服务号「集智斑图」推送论文信息。扫描下方二维码即可一键订阅:

推荐阅读
点击“阅读原文”,追踪复杂科学顶刊论文
微信扫一扫,分享到朋友圈











