老药新用治疗新冠疾病?网络医学框架加速药物研发

导语
药物研发一般需要经过一系列繁杂的过程,从虚拟筛选、临床前实验到多期临床试验等,在目前的研究范式下,一个新药往往需要投入十多年、数十亿美金;而本次新冠疫情来势如此迅猛,疫情至今仍未有公认的特效药,于是人类大多只能采用物理疏离及疫苗接种等方式,构筑免疫屏障在社会尺度上抵挡疫情,而对个体尺度的治疗则相对效果不彰。
2021年5月,PNAS 刊载了网络科学家 Albert-László Barabási 组的研究:“用于识别老药新用治疗COVID-19可能性的网络医学框架”。研究除了基于实验测得的新冠相关相互作用组数据外,还充分利用了其它复杂生物网络信息,以新冠治疗的老药新用为例,尝试建立一种高可信度的虚拟筛选方式,加速药物研发进程。
研究领域:系统生物学、网络医学、老药新用、传染病

徐恩峤 | 作者
梁金 | 审校
邓一雪 | 编辑

论文题目:
Network medicine framework for identifying drug-repurposing opportunities for COVID-19
论文链接:
https://www.pnas.org/content/118/19/e2025581118
COVID-19大流行迅猛异常,表明人类需要快速开发、测试和部署新药的方法。从头药物开发(de novo drug development)过程动辄耗时十年,显然不可行;想要节省时间,必须老药新用(drug repurposing),从已批准的药物中确定少数可能对COVID-19有治疗作用的化合物。
然而,老药新用的方法缺乏、预测单一,此次疫情中,超过1/3的临床试验集中在老药羟氯喹或氯喹上——后续结果表明几乎无疗效,但却严重挤占了其它潜在有效化合物受试的资格。如何设计一种方法高效筛选药物呢?
暴力地毯式筛选虽然可靠性更高,但成本也高,耗费时间长,效率极低 [1]。研究者转而利用多方面的复杂信息,例如分子概况、化学结构、不良反应、分子对接、电子健康记录 、通路分析、全基因组关联研究和网络扰动 [2–15]等,借助算法来排序临床试验优先级清单。其中,由于生命体本就是一个复杂系统,基于网络的方法能刻画重要信息,从而具有其它方法不具备的优势,扩大潜在药物空间。
Barabási 组的研究实现了三类网络医学老药新用算法预测候选药物。结果发现,不同方法的预测能力均较强,而提取到的特征有别。而如果在这三种预测方法之间寻求共识,采用一种多模态集成预测方法,则可以显著提高预测的准确性和可靠性 [15, 19]。
基于网络的老药新用
基于网络的老药新用
人体内所有蛋白质之间相互联系,组成一个蛋白质相互作用网络,即人类相互作用组(interactome),病毒对人体蛋白质的作用会影响这个相互作用网络。老药新用策略通常会优先考虑分子机制与新冠(COVID-19)类似的疾病,这就需要在整个人类相互作用组中,评估受新冠病毒影响的蛋白(疾病模块)与其他疾病相关蛋白的重叠。结果发现,实验确定的332个新冠病毒蛋白的宿主蛋白靶标与其他299种疾病的相关蛋白均无显著重叠[22–26]。换句话说,潜在的新冠治疗药物无法直接获得,因此可以考虑采用网络策略识别潜在药物。
研究者实施了三类基于网络的老药新用预测方法(图1)。
1) 基于 AI 的算法 [15,16]。使用图神经网络,将药物蛋白质靶点和疾病相关蛋白质映射到低维向量空间中的点,从而产生依赖于不同药物-疾病嵌入的四个预测管线(A1-A4)。
2) 基于网络扩散的算法 [17]。受扩散状态距离的启发,根据药物蛋白质靶标与新冠病毒宿主蛋白质靶标的网络相似性对药物进行排序,该算法由不同的统计方法提供支持,提供五个排名管线(D1-D5)。
3)基于网络邻近度的算法 [11]。根据新冠病毒宿主蛋白靶点与药物最近蛋白靶点之间的距离对药物进行排序,得到三个预测管线(P1-P3)。其中:P1依赖于所有药物靶点;P2去除了药物递送、代谢相关的蛋白质靶点,可以提高邻近度量的特异性;P3测试药物诱导的差异表达基因是否可以提供额外的预测能力 [27]。
统计显示,三类算法预测的药物排序结果相关性较低,表明其各自从网络中提取了不同信息。
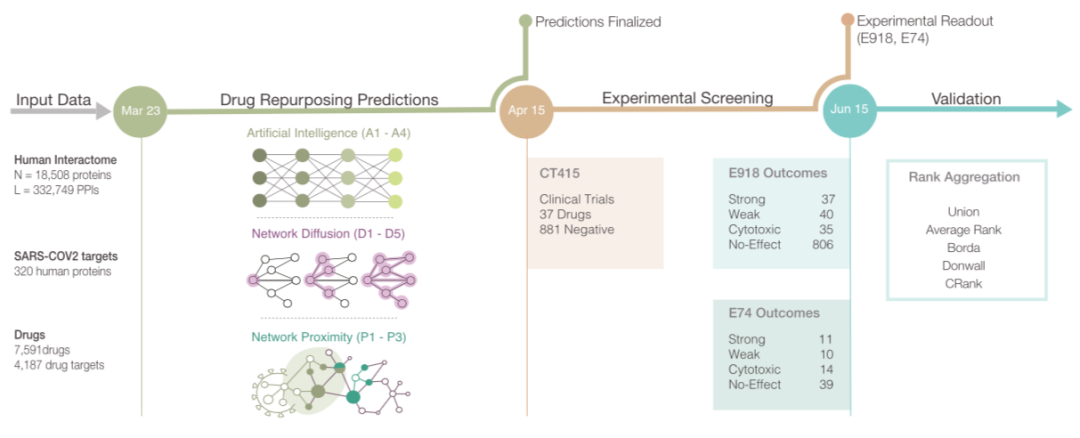
图1. 用于老药新用的网络医学框架
关键发现一:网络方法优势初显
关键发现一:网络方法优势初显
研究者实施了上面所述的共 12 个管线,来预测 DrugBank中 6340 种药物对新冠病毒的预期疗效 [27] ,并在2020年4月15日以12个排名列表的形式提取和固定预测。由于不同的管线成功预测了不同的药物子集,他们确定了所有管线都提供预测(P3因预测的药物数量明显少而除外),且其化合物在Broad Institute老药新用库[28]中可用的918种药物交集进行实验(图1)。
首先,上述预测完成后,对这918种化合物进行了试验筛选(E918 数据集),以验证它们对新冠病毒的体外抑制效果(图1)。在918种药物中,806 种对病毒感染性没有可检测的影响(记为N,No effects);35种对宿主细胞有细胞毒性(记为C,Cytotoxic);37种有很强的作用(记为S,Strong effects);40种影响较弱(记为W,Weak effects)。由于预测管线没有对体内效应的大小提供指导,研究者认为对病毒有强或弱影响的药物(S&W,77种)为阳性结果,反之则为阴性结果(N,806种)。
其次,在2020年4月15日(预测日期),研究者在134项新冠临床试验(CT415 数据集)中确定了 67 种药物。为了比较不同数据集的结果,他们将分析限制在经过试验测试的 918 种药物上,将 E918 列表上临床试验中的 37 种药物视为阳性,其余 881 种视为阴性。
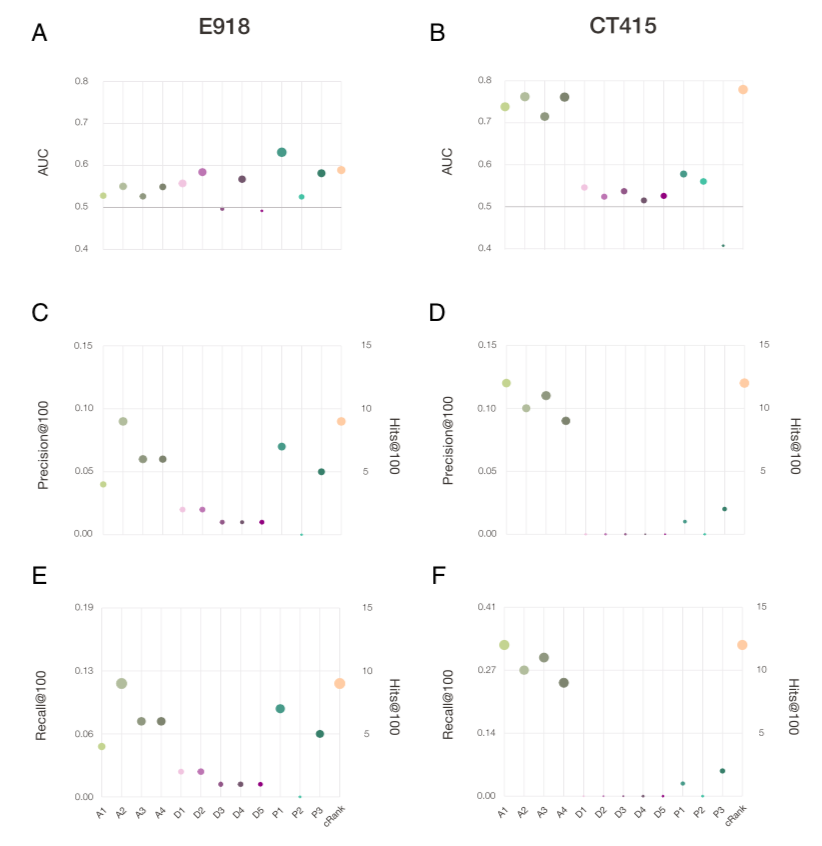
图2. 12条管线的预测性能比较
老药新用的目标是为可用药物排序,将试验工作仅限于排名靠前的化合物,从而提高效率和资源利用率。如图2所示,可以通过AUC值(取值范围为0.5~1,越接近 1 表示越准确)、排名前K的药物中的阳性结果数量(top K 精度),以及其中所有阳性结果的比例(top K召回率)这三个指标,比较12条管线的预测能力。
这就给出了第一个关键结果发现,大多数算法均具有统计学意义的预测能力,但它们在不同真实数据集上性能有别:AI管线擅于选择CT415数据集的临床试验药物,而邻近度管线对E918数据集预测更佳。不过,虽然这12条管线一起确定了前 100 名中的 22 种阳性药物,但没有一条管线为所有结果提供一致的卓越性能,这促使研究者开发一种多模态方法,以集成所有管线的预测能力。
关键发现二:不同管线集成,预测效果优异
关键发现二:不同管线集成,预测效果优异
老药新用预测模型的目标,是筛选有限数量(K)的药物参与实验,并且最大化积极结果。在最初不知道总共 Np = 12 个预测管线中的哪一个提供最佳预测能力的情况下,研究者可以从每个管线中选择前 K/Np 个药物,或者计算每种药物在 Np 管线上的平均排名。此外,排名聚合方法尝试搜索使所有管线之间预测一致数量最大化的排名[16,19]。考虑到求解的困难性,研究者实施了三种近似Kemeny共识的启发式排名聚合算法:Borda计数、Dowdall方法和CRank[16,29-31]。
结果如图3所示,平均排名的预测性能最差,落后于大多数单个管线的预测能力。合并列表和Dowdall提供了更好的结果。Borda对E918有很强的预测性能,但对CT415没有。相比之下,依赖贝叶斯因子的CRank为所有数据集和大多数 K 值提供始终如一的高预测性能。CRank在其他两个数据集上表现同样出色:手动处理的前瞻性列表 E74和2020年6月15日更新的临床试验列表C615。换句话说,研究者发现,CRank具有累积性地整合所有方法预测能力之潜力,在所有数据集中均能达到或超过单个管线的预测能力——这是研究者的第二个关键结果。
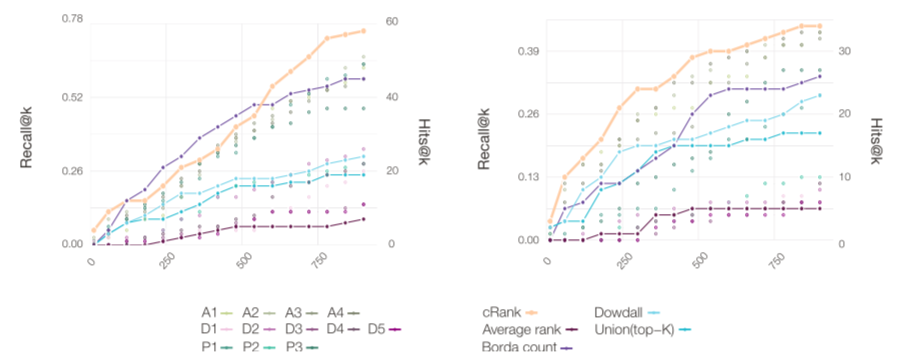
图3. 单个管线、平均排名、合并列表,以及三种排名聚合算法的预测能力比较。CRank为所有数据集和大多数 K 值提供始终如一的高预测性能。
关键发现三:
挑战传统药物研发,“网络药物”前景广阔
关键发现三:
挑战传统药物研发,“网络药物”前景广阔
目前药化领域,大多数老药新用算法依赖于分子对接,因此仅限于与病毒蛋白或病毒蛋白的宿主靶标结合的化合物 [21] ,一个很好的例子是瑞德西韦*[33, 34];而本研究还可以识别靶向其它宿主蛋白质诱导网络扰动而产生生物效应的药物,其中一些可能会改变病毒胞感染、复制的能力(图4)。完整宿主中由于潜在的蛋白质相互作用更丰富,这些药物也可能通过其他机制发挥作用,例如皮质类固醇如地塞米松[35]的抗炎作用,这只能在动物实验或临床试验中进行评估。
*注:瑞德西韦(remdesivir)在疫情初期曾被认为很可能成为新冠治疗特效药,国内曾翻译为“人民的希望”。其通过直接抑制病毒RNA聚合酶起效,原被尝试于治疗同为RNA病毒的埃博拉病毒,但目前不同临床结果却是褒贬不一。
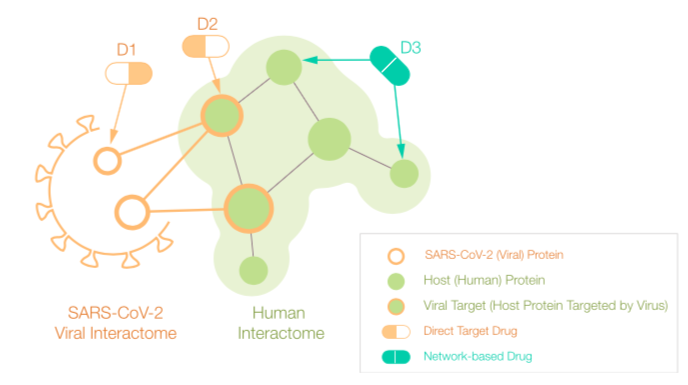
研究者发现,在 77 种 S&W 药物中,只有一种直接靶向病毒蛋白结合靶点。换句话说,在实验筛选中显示出疗效的 77 种药物中,有 76 种是“网络药物”,它们通过扰乱宿主亚细胞网络来实现治疗效果——这是研究者的第三个关键发现。
事实上,网络药物不直接针对病毒蛋白或其宿主靶标,因此针对这些蛋白的分子对接显然无效;然而,本研究通过网络方法成功地确定这些化合物纳入临床的优先级。除了老药新用的任务,网络数据还可以大幅提升药靶预测、药物联用等多种任务的预测效果(参看《Physics Reports计算网络生物学长文综述:数据、模型和应用 》);中医药由其整体观思想引导,在这一块其实出发更早,上世纪就发展了说明中药多成分、多靶点协同作用的“霰弹理论”,随后也涌现了一批重要的网络药理学研究(《生物网络 | 集智百科》中有提及)。
总之,研究者确实发现,S&W 药物靶向位于新冠疾病模块网络附近的相互作用组邻域,这可能解释了它们的抗病毒能力,并提示了网络方法的重要前景。
药物研发的算法工具集
药物研发的算法工具集
药物研发流程是循序渐进的,随着证据等级不断向上,从细胞、动物实验,到多期临床试验。有研究用VeroE6细胞(一种非人灵长类动物细胞)对12000种化合物进行体外筛选 [1],鉴定了100种抑制病毒感染的化合物,然而其中只有 39% 获得了美国食品和药物管理局 (FDA) 的批准,其余的处于临床前阶段,距离患者使用还需要数年时间。相比之下,这里优先筛选的 918 种药物中有96%获得了FDA批准,可以迅速进入临床试验。另外,暴力筛选的0.8%的命中率远低于本研究,如CRank在E918数据集的前100种药物中有9%命中率,CRank的前 800 种药物包含 77 种 S&W 药物中的 58 种(图3)。而通过专业知识和数据校正可进一步提高命中率。
还有一点值得注意的是,尽管传统药物研发流程中必定包括一定比重的临床前实验(正如本研究采用E918数据集作为ground truth),但是由于细胞、动物与人体差异巨大,其蛋白质相互作用网络不同且通常更加简化,潜在有效的药物很可能会在此类传统流程中被筛除(此类“ground truth”不一定比模拟靠谱)。而本研究使用了人类相互作用组,因此尽管是计算机模拟,在某些层面实际反而更加接近真实人体的情形。那么到底是否应该仅用此类实验去判定虚拟筛选的效果呢?这显然将对传统的药物研发流程提出挑战。
总而言之,这里介绍的方法学进步不仅为新冠提供了潜在的候选药物,而且提供了一个有原理保障的算法工具集,用于传统从头研发因金钱和时间成本而无能为力之处。
参考文献
(参考文献可上下滑动查看)
网络科学集智课堂第三期报名中
从数学建模到多学科应用
网络科学集智课堂第三期报名中
从数学建模到多学科应用
为什么新冠病毒容易在大城市间快速传播?为什么个别电站故障可以引起大范围停电?为什么手机芯片会带来国家之间的经济博弈?怎样识别社交媒体上的关键信息节点?怎样辨别社群组织内部隐藏的关键人物?怎样判定企业在上下游经济体系中的位置?……一系列真实世界问题的挑战,催生了复杂网络研究的快速兴起。网络科学正在成为21世纪的新通识。
集智学园特邀陈关荣、樊瑛、周进、李翔、张江、闫小勇、刘宗华、石川、虞文武、赵海兴、史定华等网络科学专家作为导师,自10月16日起开展系列在线课程,介绍复杂网络的建模与应用以及相关课题。欢迎希望进入网络科学领域、提高网络分析能力、与一线专家探讨问题的朋友报名参加!

推荐阅读
点击“阅读原文”,报名课程
微信扫一扫,分享到朋友圈











