研究速递:深度学习引入适应性,提升蛋白质结构预测能力

摘要
元动力学和伞形采样等改进的采样方法,已经成为探索分子和材料构型空间的重要工具。与此同时,它们长期以来面临着一些问题,如在处理大量集合变量或具有高能量势垒的系统时效率低下。在这里,我们表明,通过聚类和适应性调整技术,强化动力学算法(reinforced dynamics, RiD)可有效地探索含大量集合变量或高能量势垒的构型空间和能量景观。我们通过研究各种具有代表性和挑战性的例子来说明这一点。首先,我们通过与其它方法的比较,证明了适应性 RiD 的有效性,并构造了能量势垒大于8kcal mol-1的九维(9D)能量景观。然后,我们使用18个集合变量研究了蛋白质 chignolin 的折叠。在这种情况下,观察到的折叠和展开速率均为4.30 μs-1。最后,我们提出了一种基于 RiD 的蛋白质结构修正工具。该工具使我们能够有效使用超过 100 个集合变量来探索蛋白质结构的能量景观,并使其GDA-HA 分数相比于初始结果平均提高了14.6分。
研究领域:分子动力学,能量景观,蛋白质结构预测,适应性算法

张澳 | 作者
邓一雪 | 编辑

论文题目:
Efficient sampling of high-dimensional free energy landscapes
using adaptive reinforced dynamics
论文地址:
https://www.nature.com/articles/s43588-021-00173-1
1. 生物分子模拟难以逾越的势垒
1. 生物分子模拟难以逾越的势垒
分子动力学模拟是对生物分子结构及其动力学进行建模的重要工具。然而,生物分子的能量景观存在众多能量势垒,克服能量势垒又是小概率事件。因此对生物分子的动力学模拟仅能维持在较小的时间尺度。具体而言,生物分子的能量景观是集合变量(collective variables,即分子动力学模拟过程中可测量的结构参数,如到结合位点或膜的距离)所组成的高维构型空间的能量函数图。当存在能量势垒时,构型空间被其分隔(图1B),蒙特卡洛采样可能无法探索足够的构型空间以精确重构能量景观。 相比之下,不存在能量势垒的景观结构(图1A)则有利于采样。
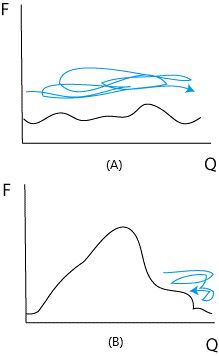
为重构存在能量势垒的景观,诸如元动力学(metadynamics)、伞形采样(umbrella sampling)等经过改进的采样方法被提出。其主要改进是引入加权函数,给势能添加偏置势(biasing potential,为系统一个或多个集合变量的函数),从而抵消能量势垒的影响。而近年来快速发展的机器学习为采样方法提供了更强有力的工具,核函数、深度神经网络等方法能够作为逼近器(approximator)有效逼近高维函数,从而助力于重构高维能量景观。不同的采样方法仅在逼近器参数优化及其实时改进采样的方式上有所区别,通常以过往势能的平均值优化逼近器参数。
2. 适应性RiD算法突破势垒
2. 适应性RiD算法突破势垒
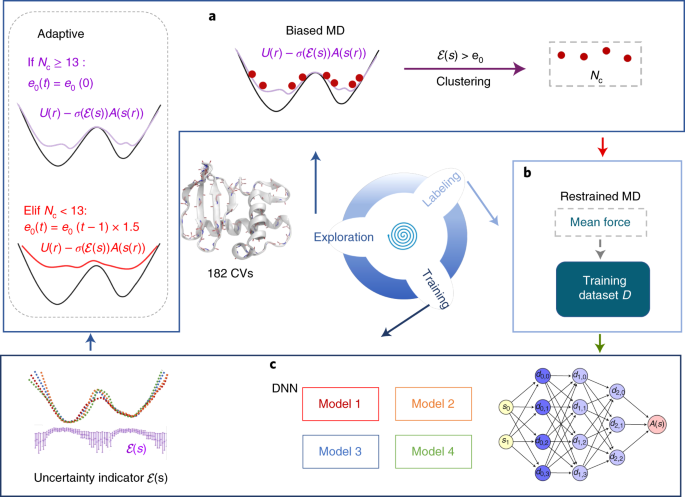
Nature Computational Science近日发表的一篇论文中,研究者提出了一种具备适应性的深度神经网络方法——reinforced dynamics算法(后文简称RiD),该方法能够有效探索大量集合变量或高能量势垒。这一算法是传统 RiD 算法的改进版。RiD 算法采用不确定性指标来决定在何处对势能进行偏置,以及当集合变量为何值时需计算平均势能。当集合变量的数目小于 20 时,RiD 算法能够很好地探索构型空间,但若变量数目进一步增加,其效率会迅速降低。究其原因,当构型空间维度变高时,算法会被困在局部最小值(deep local minima)。而脱困存在两个难点:(1)探索局部最小值周围区域的可能性随着维度增高而降低;(2)偏置机制过于僵化。
适应性 RiD 算法基于两个优势,得以脱困于局部最小值,即(1)采用聚类算法对用于标记的构型分类,减少表征未探索构型空间所需的构型数量;(2)同样利用聚类算法,适应性地迭代不确定指标及偏置势。具体而言,每次迭代包含三个步骤(图2):探索、标记和训练。在探索步骤中,通过集合变量空间中定义的偏置势优化构型空间的采样。其中是否添加偏置势取决于不确定指标的值,该值被定义为来自 DNN 模型集间预测值的标准差。首先根据不确定性指标选择所探索的构型空间,然后通过聚类选择预测误差较大的构型空间子集。在标记步骤中,计算每个所选构型空间聚类的平均能量作为其标记。在训练步骤中,以标记值训练一组初始参数随机且独立的 DNN 模型集。随后,通过计算DNN模型集间的标准差来计算不确定指标,进一步优化构型空间的采样,周而复始。
3. 高效采样进一步雕琢蛋白质结构
3. 高效采样进一步雕琢蛋白质结构
适应性 RiD 算法能够有力推进蛋白质结构采样、环区优化以及结构修正等工作。在国际蛋白质结构预测竞赛CASP13中三个典型靶点(R0974s1,R0986s1,R1002-D2)上,相比于AlphaFold2预测的初始结果,经适应性 RiD 算法修正后的结果 GDT-HA(Global Distance Test-High Accuracy,以0-100分量化预测结果与靶点结构的相似度)平均提升了14.6分,效果远超其余修正算法。
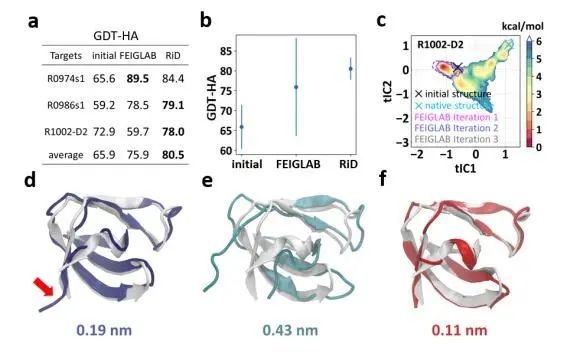
生物分子能量景观的高效采样是分子动力学的关键问题,该研究开发的适应性 RiD 算法能够处理超过100个集合变量,并能克服高能量势垒,出色地解决了这一问题,进而为药物设计、材料优化等一系列重要领域提供了突破的可能性。
复杂科学最新论文
集智斑图顶刊论文速递栏目上线以来,持续收录来自Nature、Science等顶刊的最新论文,追踪复杂系统、网络科学、计算社会科学等领域的前沿进展。现在正式推出订阅功能,每周通过微信服务号「集智斑图」推送论文信息。扫描下方二维码即可一键订阅:

推荐阅读
-
生物物理前沿:量化细胞内生化信号分子动力学编码的信息积累 -
PNAS前沿:系统何时走向崩溃?深度学习算法帮助预警临界点 -
蛋白质的动力学和进化之间的对应关系:两个不同时间尺度下的相同故事 -
《张江·复杂科学前沿27讲》完整上线! -
成为集智VIP,解锁全站课程/读书会 -
加入集智,一起复杂!
点击“阅读原文”,追踪复杂科学顶刊论文
微信扫一扫,分享到朋友圈











