新冠疫情好似一场世界大战,病毒演化与人类抗疫手段的发展在上演一出特别的军备竞赛。Alpha、Beta、Delta以及最新的Omicron变异毒株不断冲击着人类的抗疫战线。面对如此战况,我们如何把握主动?是否有可能预测新冠病毒的演化?Fred Hutchinson 癌症研究中心的研究人员借助深度突变扫描技术,勾绘出冠状病毒刺突蛋白中受体结合区的适应度景观,尝试理解病毒如何通过演化加强传染性并躲避免疫系统的侦察,以此为后续疫苗的研发指明道路。
研究领域:传染病,适应度景观,协同演化
Carrie Arnold | 作者
赵雨亭 | 译者
张澳、梁金 | 审校
邓一雪 | 编辑
2019 年秋冬,现代历史上最大的一场演化生物学实验开始了。一种冠状病毒获得了生活在人类体内而非蝙蝠或其他哺乳动物体内的能力,它进一步适应以高效地从一个人传播到另一个人,甚至在身体的防御能力启动之前就开始传播。但病毒的演化游戏并没有就此停止,研究人员不得不用一个个希腊字母来命名 SARS-CoV-2 的新变种。
世界各地的研究人员正试图更详细地了解病毒的演化过程,特别是 SARS-CoV-2 的突变如何改变其在人类之间传播的能力。“今天适应良好的病毒明天可能会因为宿主产生抗药性而逐渐消亡,所以病毒必须找到一种新的方法来感染宿主。这推动了自我提升的更新。”加州大学圣地亚哥分校的演化生物学家 Justin Meyer 表示。
尽管不断变化的疫情造成了大量人员伤亡,但观察病毒在全球范围内传播的大量科学数据具有指导意义。牛津大学大数据研究所的统计遗传学家 Luca Ferretti 表示,COVID 为研究人员提供了珍贵的演化实例。
准确预测病毒下一步的发展是痴心妄想,但世界各地的病毒学家一直在深入研究 SARS-CoV-2 中哪些成分最容易演化,哪些关键蛋白质元素的改变会影响其生存 [1]。这些信息可以为研发更好、更持久的疫苗指明道路。其它研究则聚焦于病毒对单克隆抗体疗法(monoclonal antibody therapies,一种治疗新冠重症患者的疗法)产生耐药性的方式[2]。这项工作还指出,特定变体组合一旦广泛传播,就可能引发新一轮疫情。这些变体除了传播速度快之外,还尤为擅长攻击人体的免疫系统[3]。
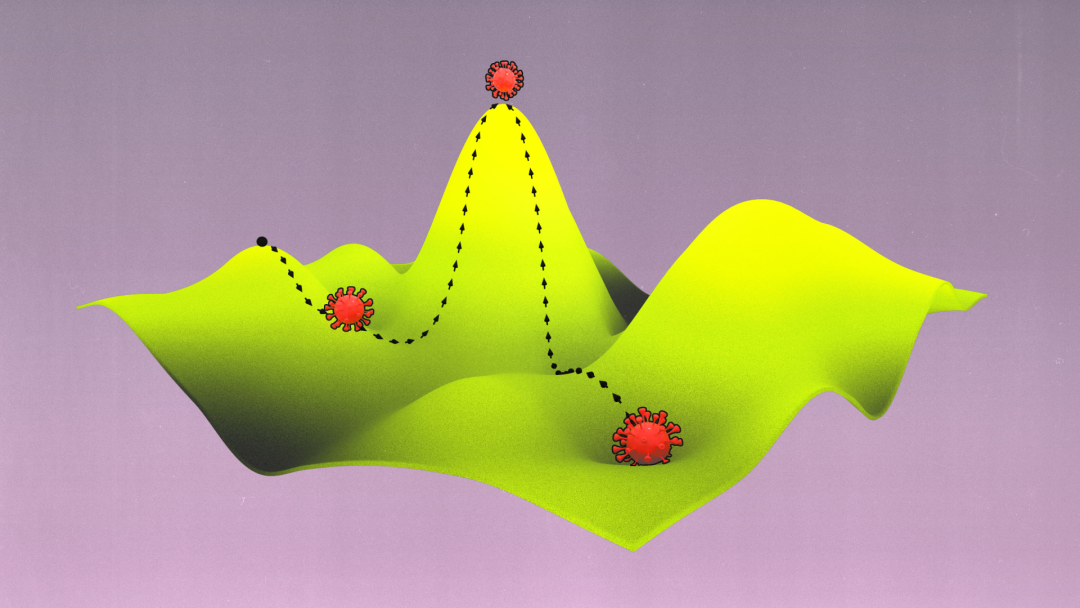
研究人员已经能够通过使用现代技术重新审视近一个世纪前提出的概念——适应度景观(fitness/adaptive landscape)——来做出以上发现。他们可以用适应度来量化病毒基因组变化与其复制和感染新宿主能力之间的关系。代表这种关系的景观图可以帮助重建病毒的历史,甚至还可以预测病毒的未来。
对于伦敦帝国理工学院的分子演化生物学家 Tobias Warnecke 来说,适应度景观是将基因型与表型联系起来的宝贵方式。他表示,通过定量发掘基因的潜力,研究人员可以探索两个突变如何共同影响一个性状,以及第三个突变的存在如何影响这两个突变。“通过这种方式,可以查看许多不同的基因型组合,看看它如何影响自己感兴趣的东西。”
适应度景观的价值不仅限于比较基因组或蛋白质的少量变化。现代实验技术开发了一种称为深度突变扫描(deep mutational scanning)的技术,利用该技术,研究人员能够对自然选择进行小规模实验,并同时比较数万个突变的适应度。该过程可能有助于揭示病毒变体间无法预料的相互作用,并且尝试找出可能对人类构成新威胁的病毒未来演化的路径。
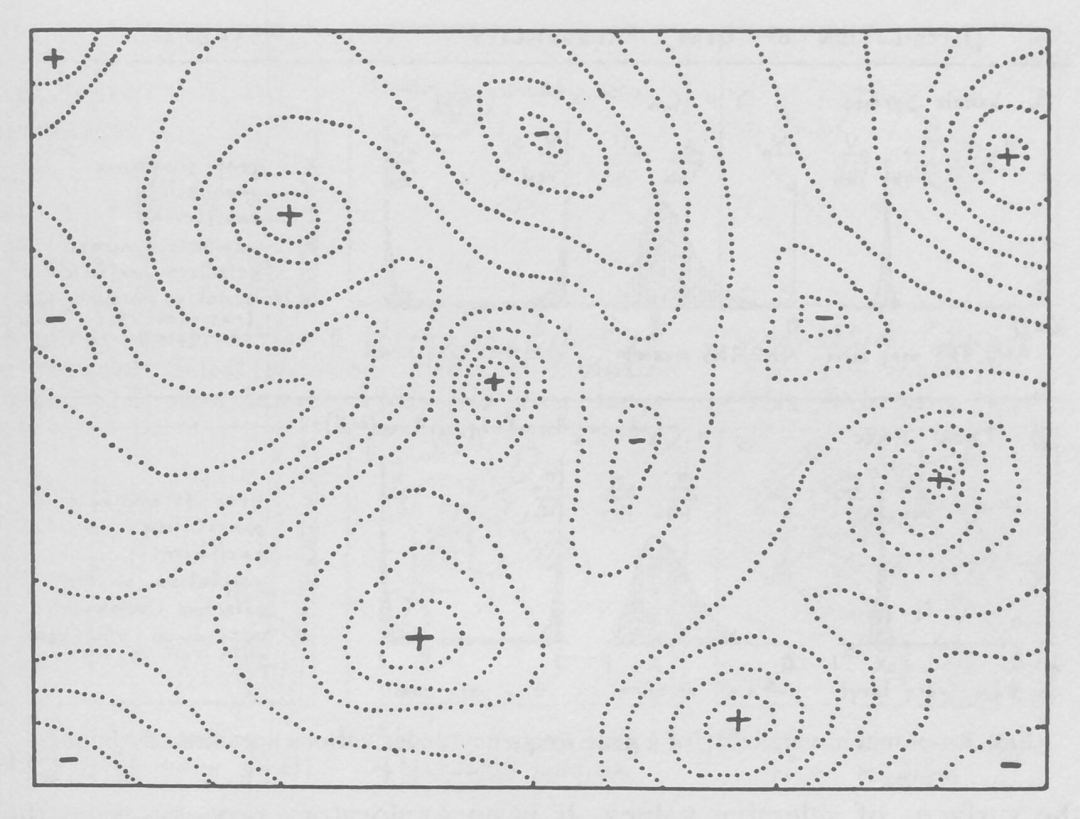
在《物种起源》中,查尔斯·达尔文写道,自然选择是“保持有利的个体差异和变异,消灭有害的个体差异和变异”的结果。在基因的概念出现之前,生物学家只能试图想象有机体微小、可遗传的变化如何影响繁殖。美国生物学家 Sewall Wright 的工作首次实现了该想法。在1932 的《第六届国际遗传学大会论文集》上发表的论文中,Wright 开创性地使用手绘图来表明有机体如何穿梭于“几乎无限大的可能变异领域,并在自然选择下找到生存之路”。
Wright 指出,可视化大量可能的线性分子(如 DNA 或肽)变体的一种方法,是将每种可能性视为空间中一个特定的点。分子演化即为初始变体点和最终变体点之间的路径,该路径途经中间变体的所有点。
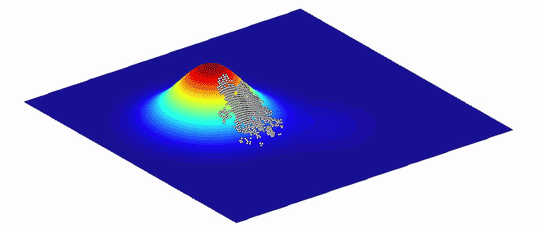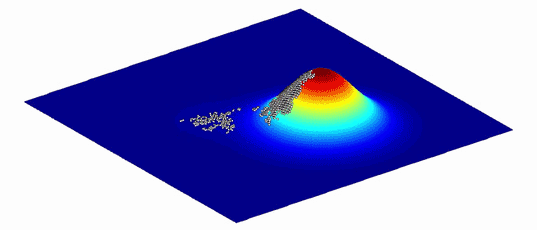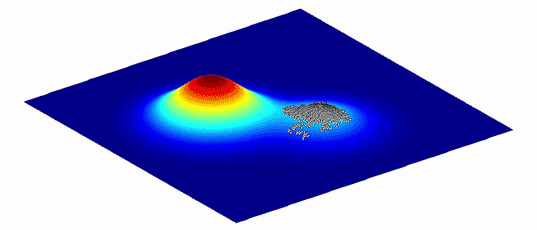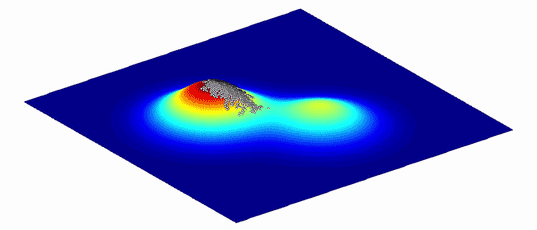
为了帮助理解这些变体的复杂图形以及它们之间的演化路径,Wright 将其表示为更直观的仅有两个或三个维度的“适应度景观”。平面轴绘制 DNA(基因型)或物理特征(表型)的变异;两个变体越相似,它们在平面上的位置就越近。纵轴衡量变异对演化适应度的影响。提高生物体存活几率的变体,无论是通过增加其存活后代还是改善其蛋白质的功能,均集中在适应度景观的高峰,而降低生物体存活几率的变异则被困在山谷中。
图1. Sewall Wright 为其 1932 年的开创性论文绘制的一幅插图,代表了适应度景观可能的模样。该平面代表所有可能变体的集合。其中虚线表示具有相似适应度的变体,加号表示景观的高峰,减号表示景观的山谷。| 来源:The University of Chicago Library and permissions granted from Genetics Society of America
密歇根大学医学院的演化生物学家 Adam Lauring 解释,由此产生的景观具有独特的拓扑。如果映射的变体对适应度的影响没有太大差异,那么景观看起来就像平原一样平坦。对人类健康影响巨大的变体则在景观中创造了类似珠穆朗玛峰一样的高峰。自然选择偏爱高峰所对应的变体:物种平均基因型或表型应该通过在高峰间移动来演化,并且演化最好是沿着高峰间的山脊而非穿过山谷。(具有不同基因型的孤立亚群也可以帮助一个物种找到跨越鸿沟的方式。)
“如果你移动几英尺,就会摔倒,再次站起来会变得非常困难,移动的途径会越来越少。”Lauring表示。
“这个理论非常简单。研究人员只需要知道物种的基因型,然后测量其对应的适应度,便可以预测任何可能发生的事情。”在瑞士伯尔尼大学研究演化动力学的 Claudia Bank 表示。但将理论付诸实践就是另一回事了。
一个复杂的问题是,无论是针对 SARS-CoV-2 还是人类,适应度景观都不是一成不变的。对于生物体而言,一种能够使其消化新食物、但同时也降低生长速度的变异,既可能是救命稻草,也可能是致命障碍。变异对演化适应度的影响取决于生物体所处的环境。当环境发生变化时,适应度景观也会发生变化。“不同的变异影响不同,这取决于生物体当前的环境。” Lauring表示。
图2. 由于环境不断变化,适应度景观也总是在不断变化。生物种群追随高峰而演化。| 来源:Randy Olson & Bjørn Østman
绘制适应度景观也是一个数学挑战。长度只有 100 个氨基酸的小蛋白质会有 20100 种可能的变体——比宇宙中原子的数量还要多。很难想象,更不用说去计算真实蛋白质适应度景观的复杂拓扑结构及其可能路径的数量。因此,几十年来,适应度景观只是概念上的辅助,而非具体测量的工具。直到最近,借助先进的计算能力和改进的分子生物学技术,研究人员才能够开始为单个蛋白质和细菌、病毒等简单生物体绘制定量景观。
细菌和病毒几乎是绘制适应度景观的理想对象。在试管中生长至数百万或数十亿的数量,每个细菌细胞或病毒颗粒都可以从描适应度景观的巨大变体库中携带一个突变。它们极短(几小时或几天)的代际差,也使研究人员能够更快地识别新的突变。大多数使用 RNA 作为其遗传物质的病毒,包括 HIV 和丙型肝炎病毒(HCV),也很容易发生突变,因为复制其基因组的 RNA 聚合酶不会像 DNA 聚合酶那样有效地校对拷贝。
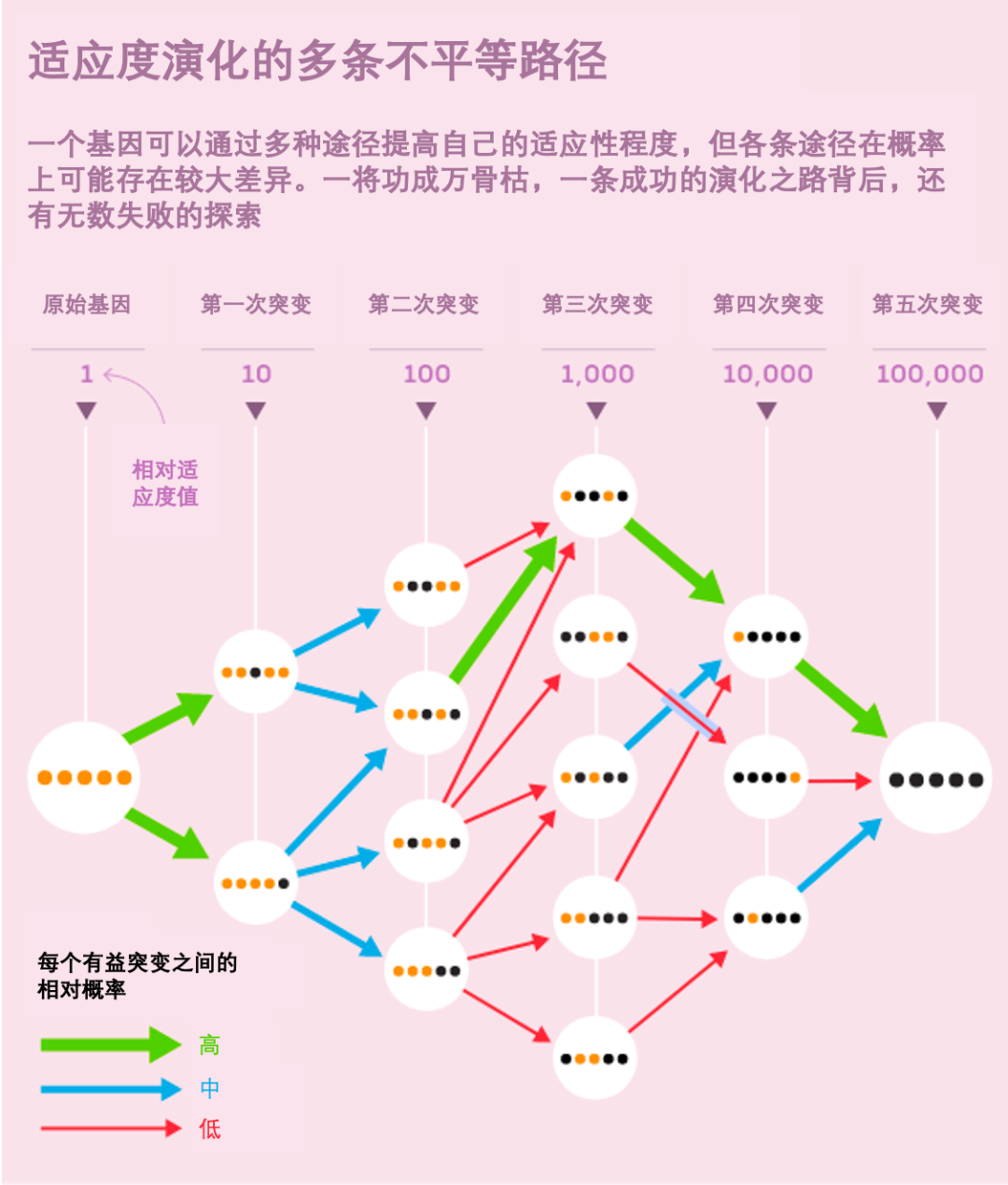
研究人员首先发现,尽管适应度景观很复杂,但生物体通常仅处于少数适应度峰值,并且其间的路径数也受限。2006 年发表于 Science 的一篇论文仔细研究了一种叫做 β-内酰胺酶的蛋白质,它可以使青霉素等抗生素失活 [4]。β-内酰胺酶中五个单核苷酸突变的联合作用可以使其抗生素耐药性增加 100000 倍。时任哈佛大学演化生物学博士后、现任布朗大学实验室负责人的 Daniel Weinreich 与他的同事指出,该基因的演化可能以 120 条路径来累积所有五种变体。
图3. β-内酰胺酶可能以120条路径来积累变体。| 来源:Samuel Velasco/Quanta Magazine; doi.org/10.1126/science.1123539
然而,当研究人员在实验室中创建和测试中间变体时,他们发现有 102 条路径在自然选择下是不可能存在的,因为它们产生了有缺陷或不完整的蛋白质。随后他们发现许多剩余的组合未能改善抗生素耐药性,可能的演化路径进一步减少。对此,他们表示,“生命的蛋白质演化途径可能在很大程度上是可复制的,甚至是可预测的。”
但即使是预测最小的病毒或蛋白质的演化轨迹,也需要对其适应度环境有详尽的了解,而这很难达成。从历史上看,科学家仅能一次产生一个核苷酸或氨基酸的变体,然后纯化蛋白质变体并评估其功能。而检查多个可能的变体通常是不实际的。
深度突变扫描技术 [5] 的发展改变了这一切。这种技术允许科学家一次性生成数以万计的变体,然后让所有变体相互竞争以确定它们的相对适应度。
研究人员首先创建了一个可以克隆到培养细胞中的变异基因库。由基因编码的蛋白质,其活性与一些生化功能有关。在实验室中对生化功能施加选择,可增加适应度最高、活性最高的蛋白质,而活性低的蛋白质将消失。通过高通量 DNA 测序,研究人员可以统计每个变体的数量,以定量测量其多代的表现。
“这是一种刻画变体影响的有效方法,”伦敦 Warnecke 实验室的研究员 Valerie Soo 表示。
对于易突变的 RNA 病毒,科学家甚至不必在实验室中产生变体——易出错的基因组复制流程会引入突变,自然产生变体。病毒的数以百万计的拷贝,每个都与其相邻病毒略有不同,从而产生病毒学家所定义的突变群(mutant swarm),这是自然选择促进演化的原材料。
“微生物繁殖速度如此之快,以至于演化每天都在发生。研究人员实际上可以实时监测演化,”法国蒙彼利埃 MIVEGEC 实验室的演化生态学家 Samuel Alizon 表示。
研究人员发现,突变群中的突变很少能被传播给新宿主,特别是当感染仅需少量病毒时。其中一些传播纯粹是偶然,即变体在正确的时间出现在正确的位置。但通过勾绘适应度景观,研究人员可以尝试找出为什么某些变体的传播频率远高于其他变体,加州大学旧金山分校的病毒学家 Raul Andino-Pavlovsky 表示。“病毒不仅需要能够产生多样性,而且还必须能够容忍这种多样性,能承受变化的病毒很可能是一种适应性更强的病毒。”
图4. Fred Hutchinson 癌症研究中心的研究人员 Tyler Starr(左)和 Jesse Bloom(右)研究了 SARS-CoV-2 刺突蛋白的一个关键结构域,以找出其引起免疫系统注意的部分。| 来源:Tyler Starr & Jesse Bloom
根据演化生物学家 Tyler Starr 的说法,不管是在概念上还是定量化上,适应度景观都是描述慢性或持续感染中,病毒如何规避宿主免疫系统中和作用的完美方式。他加入 Fred Hutchinson 癌症研究中心 Jesse Bloom 实验室,就是为了研究 HIV 如何在感染过程中与患者体内的抗体免疫共同演化。他的目标是了解病毒和免疫系统之间的这种演化军备竞赛如何产生具有保护性的抗体,这可以帮助科学家开发关注病毒中恒定部分的 HIV 疫苗。
但 Starr 刚开始他的 HIV 研究工作,另一种病毒就抢走了他以及全世界的注意力。
随着 SARS-CoV-2 开始在全球传播, Starr 和 Bloom 意识到,适应度景观为研究这种新型病原体提供了一种有用的方法。适应度景观能够确定病毒蛋白质中的关键因子,以及病毒可以承受的变化。
最初,对 SARS-CoV-2 进行测序的科学家并没有注意到太多的遗传变异。尽管冠状病毒以较易出错的 RNA 聚合酶来复制遗传物质,但 SARS-CoV-2 有另一种蛋白质校对拷贝。因此,研究人员并未预期 SARS-CoV-2 会像流感或 HIV 般存在大量变体。
Bloom 和 Starr 知道,在最强烈的演化压力下,刺突蛋白(spike protein)将成为冠状病毒的一部分。因为刺突蛋白是免疫系统在识别上最敏感的物质,也是病毒用来侵入人体细胞的物质。然而,刺突蛋白含有1273个氨基酸,如此大的规模使其无法通过适应度景观进行快速评估。因此, Starr 决定专注于刺突蛋白的一个部分,即仅包含几百个氨基酸的受体结合区(receptor binding domain)。后者只有几百个氨基酸,是一个更容易解决的问题。
Starr 使用深度突变扫描创建了 4000 种不同的受体结合区变体。他评估了它们与人类 ACE2 蛋白(病毒进入细胞所选择的分子“锁”)结合并被免疫系统识别的能力 [6]。如果 SARS-CoV-2 不能承受其受体结合区的变异,Starr 将会看到免疫识别或 ACE2 结合功能受到变体的严重损害。
SARS-CoV-2 刺突蛋白(底部)与细胞受体结合的受体结合区模型,这种相互作用使病毒能够进入细胞。蛋白质的红色区域不能轻易演化,因为其突变会阻止蛋白质工作。| 来源:Tyler Starr
但这并不是实际发生的事情。“受体结合区有许多不同的变体,它们实际上提高了亲和力,” Starr表示。“这看起来像是一个非常包容的结构域,具有很高的演化能力。然而,当时的想法是冠状病毒不会以抗原方式演化。病毒基因组可能很稳定。”
虽然受体结合区能够承受比预期更多的变异,但并不是所有的刺突蛋白都可以。Starr 表示,刺突蛋白的这些部分因此可能是新疫苗和单克隆抗体的良好靶标,因为它们不太可能随着时间推移发生突变。
Starr 表示,当他们于 2020 年 6 月首次在 biorxiv.org 预印本服务器上发布这些结果时,敲响了一个巨大的警钟,这是 SARS-CoV-2 比人们想象的更易变异的最初迹象之一。现在 Starr 和 Bloom 正在重复他们对 alpha、beta、gamma、delta 和 omicron 变体的深度突变扫描实验,以获得关于它们的受体结合区的类似见解。
Starr、Bloom 及其同事还创建了一个不涉及 ACE2 结合体的受体结合区所有可能突变的图谱。他们在 2021 年 1 月发表于 Science 的工作 [7],确定了该结构域可能规避单克隆抗体疗法中和作用的可能突变。他们的工作还识别了在感染 SARS-CoV-2 后 150 天的免疫功能低下的个体中出现的几种突变。当病人在第 145 天接受单克隆抗体治疗时,病毒已经对市面上可用的药品产生了耐药性。对 Starr 来说,这表明这些具有治疗效果的单克隆抗体可能会随着时间的推移而变得不那么有效,无论是在单个患者体内,还是面对更普遍的病毒突变时。
此外,正如 Starr、Bloom 和他们的同事去年夏天在 Nature Communications [8] 上所描述的那样,几种普遍的突变都可以帮助 SARS-CoV-2 规避免疫系统针对受体结合区中最具针对性的部分的一些抗体。到目前为止,还没有病毒谱系演化为具有所有这三种突变。“然而,研究人员认为这种变体的出现将是一个令人担忧的发展,应该密切关注。”他们写道。
2019 年底首次出现 SARS-CoV-2 的世界与当今世界不同。病毒产生大量拷贝并在个体间传播的能力无疑是其在疫情初期取得成功的关键。然而,随着通过疫苗接种和自然获得性感染免疫的人数增加,病毒将承受更大的压力来规避免疫反应。Lauring 表示,许多突变都需要权衡利弊,SARS-CoV-2 也不例外。能够帮助病毒规避免疫,但却减少其传播的突变可能在 2020 年初不受青睐,但现在则不同。
Lauring表示:“我们人类是病毒生存的环境,如果我们改变,适应度景观就会改变。”
[1] Huang, Y., Yang, C., Xu, Xf. et al. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol Sin 41, 1141–1149 (2020). https://doi.org/10.1038/s41401-020-0485-4
[2] https://www.science.org/doi/10.1126/science.abf9302
[3] Greaney, A.J., Starr, T.N., Barnes, C.O. et al. Mapping mutations to the SARS-CoV-2 RBD that escape binding by different classes of antibodies. Nat Commun 12, 4196 (2021). https://doi.org/10.1038/s41467-021-24435-8
[4] https://www.science.org/doi/10.1126/science.1123539
[5] Carlos L. Araya, Douglas M. Fowler. Deep mutational scanning: assessing protein function on a massive scale. Trends in Biotechnology, Volume 29, Issue 9,
2011, Pages 435-442, ISSN 0167-7799. https://doi.org/10.1016/j.tibtech.2011.04.003.
[6] Tyler N. Starr, et al. Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell, VOLUME 182, ISSUE 5, P1295-1310.E20, SEPTEMBER 03, 2020. DOI:https://doi.org/10.1016/j.cell.2020.08.012
[7] https://www.science.org/doi/10.1126/science.abf9302
[8] Greaney, A.J., Starr, T.N., Barnes, C.O. et al. Mapping mutations to the SARS-CoV-2 RBD that escape binding by different classes of antibodies. Nat Commun 12, 4196 (2021). https://doi.org/10.1038/s41467-021-24435-8
https://www.quantamagazine.org/evolution-landscapes-predict-whats-next-for-covid-virus-20220111/
集智斑图顶刊论文速递栏目上线以来,持续收录来自Nature、Science等顶刊的最新论文,追踪复杂系统、网络科学、计算社会科学等领域的前沿进展。现在正式推出订阅功能,每周通过微信服务号「集智斑图」推送论文信息。扫描下方二维码即可一键订阅:

点击“阅读原文”,追踪复杂科学顶刊论文