关键词:多组织基因表达插补,转录组学,超图,图表示学习,注意力机制,编码器-解码器
论文题目:Hypergraph factorization for multi-tissue gene expression imputation
斑图地址:https://pattern.swarma.org/paper/dd285798-24f5-11ee-931d-0242ac17000d
论文地址:https://www.nature.com/articles/s42256-023-00684-8
多组织插补(Multi-tissue imputation)是转录组学的核心技术之一,用于处理在多组织基因表达数据中的缺失值,进而帮助阐明调节各种发育和生理过程的机制。插补算法利用其他相关组织的信息来预测缺失的基因表达值。例如,如果肝脏组织的基因表达数据中有缺失值,可以使用来自胰腺或肾脏等相关组织的数据来预测这些缺失值。针对基因表达的插补算法,通常涉及到统计模型或机器学习算法,从已知数据中学习基因表达模式,并预测缺失数据。但在计算上,多组织转录组插补具有挑战性,因为数据维度随着基因和组织的规模变化而快速变化,通常导致模型过参数化。
最新发表在 Nature Machine Intelligence 的论文中,作者提出了一种图表示学习方法——HYFA(超图因子分解,hypergraph factorization),用于同时处理来自多个组织和多种细胞类型的基因表达数据,并预测其中的缺失值。该方法在处理大规模基因表达数据时,具有较高的计算效率和准确性。
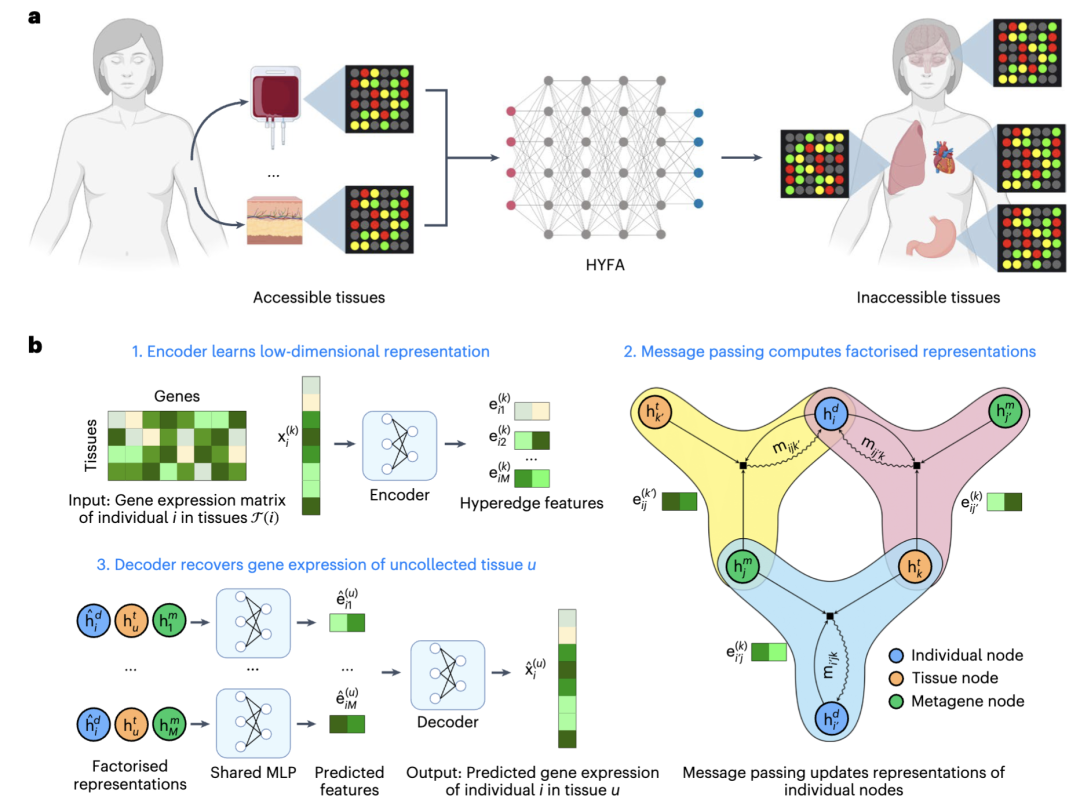
具体而言,在HYFA中,多组织基因表达被表示为一个由个体(即基因测序生物个体)、元基因(即一组或一类基因的集合)和组织(即基因表达数据的来源部位)构成的超图。其中每个节点代表一个元基因,每个超边(hyperedge)连接到一组元基因节点,代表了一个个体在特定组织中表达的基因集合。使用超图数据结构可以捕捉到基因表达数据中的复杂模式,包括基因之间的相互作用、基因在不同组织中的表达差异,以及不同个体之间的差异,进而为机器学习提供丰富上下文信息。
HYFA架构的关键部分是超图消息传递的神经网络,采用注意力机制,对不同超边赋予不同权重,然后根据权重对超边进行聚合,从而得到每个节点的新的特征表示。这种机制帮助模型确定哪些信息更重要(注意力权重反映了某个节点对其他节点的重要性),从而缓解过度压缩问题,并增强可解释性。
HYFA的另一个重要特性是它并不依赖于具体基因型,而是依赖于基因表达的模式和组织的类型,因此可以支持每个个体收集的不同的组织数量,并且可以利用组织和基因的共享调控结构(即基因/组织/个体关系的先验知识或假设),有效地进行基因表达插补。
HYFA模型还包括一个编码器和一个解码器,编码器负责将输入数据转换为低维隐空间,解码器则将隐空间数据转换回原始高维空间,便于模型处理高维数据。
实验和分析表明,该方法是参数高效的,无需大量参数,可以在较少的数据上进行有效学习。HYFA在处理基因表达数据时,不需要获取个体的基因型信息,同时可以应对数据中每个个体收集的组织数量不同的情况,因此在实际应用中更加灵活。使用HYFA插补的数据集,研究人员可以更有效地识别影响基因表达遗传位点的遗传变异。
此外,HYFA不仅可以用于单个数据集的基因表达插补,还可以用于整合多个组织和/或细胞类型的转录组数据,这有助于构建和利用大规模的转录组生物库。
图 1 本文 HYFA 模型架构示意图。其中 a为HYFA的超图结构,b 为模型的注意力机制,c 为编码-解码过程。
推荐阅读