关键词:蛋白质结构预测,错义变异分类,蛋白质语言模型,机器学习
论文题目:Accurate proteome-wide missense variant effect prediction with AlphaMissense
斑图地址:https://pattern.swarma.org/paper/f3b4b46a-58e0-11ee-88c4-0242ac17000d
论文地址:https://www.science.org/doi/full/10.1126/science.adg7492
基因组测序揭示了人类群体中广泛的遗传变异。错义变异(missense variant)是改变蛋白质氨基酸序列的遗传变异。致病性错义变异破坏蛋白质功能,降低机体适应性,而良性错义变异作用有限。
对这些变异进行分类是人类遗传学中一个重要的持续挑战。在观察到的400多万个错义变异中,估计只有2%在临床上被归类为致病性或良性,而其中绝大多数具有未知的临床意义。这限制了罕见病的诊断,以及针对潜在遗传原因的临床治疗的发展或应用。机器学习方法可以通过利用生物学数据中的模式来预测未注释的变异的致病性,从而缩小变异解释的差距。具体而言,AlphaFold 能够从蛋白质序列中准确预测蛋白质结构,可以作为预测蛋白质变异致病性的基础。
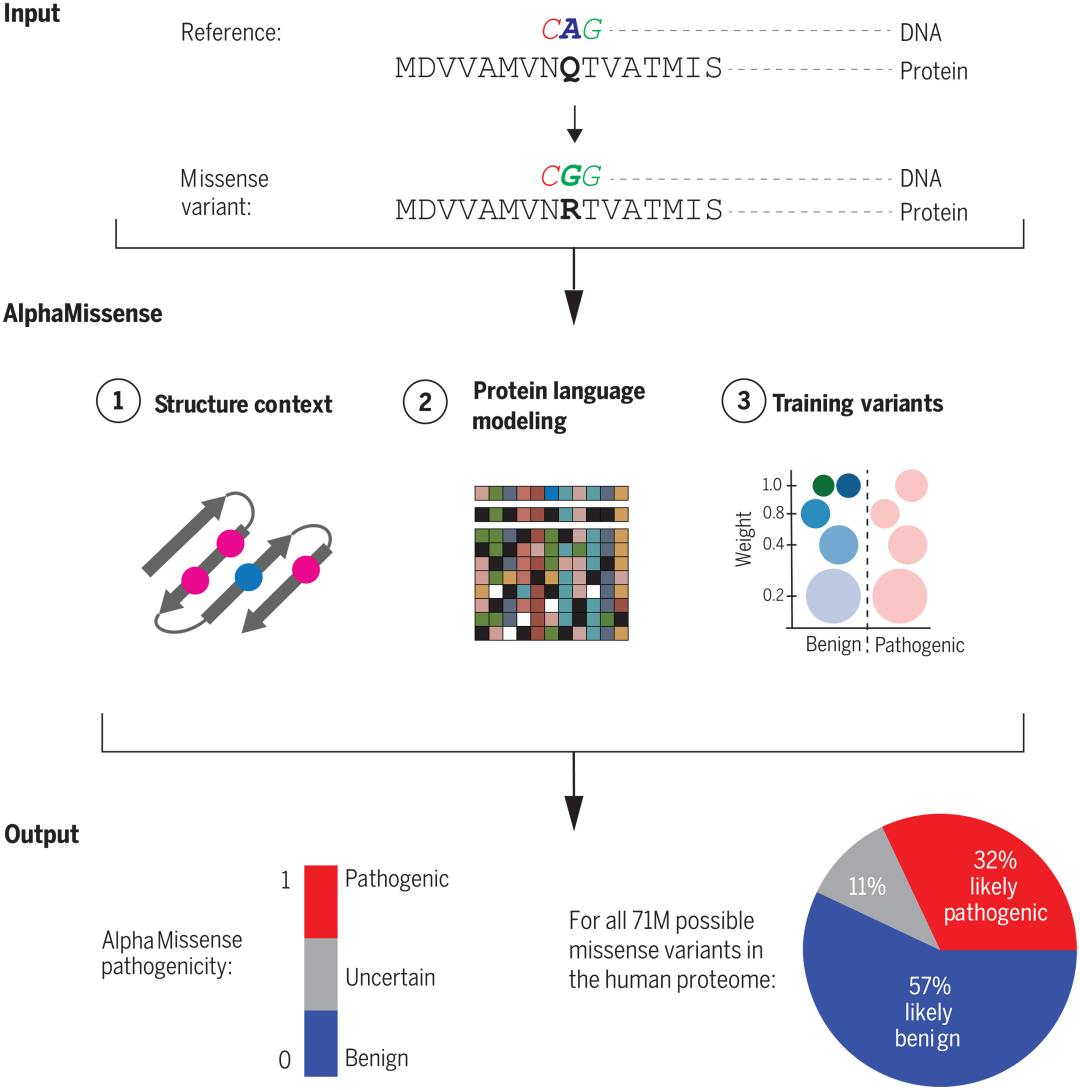
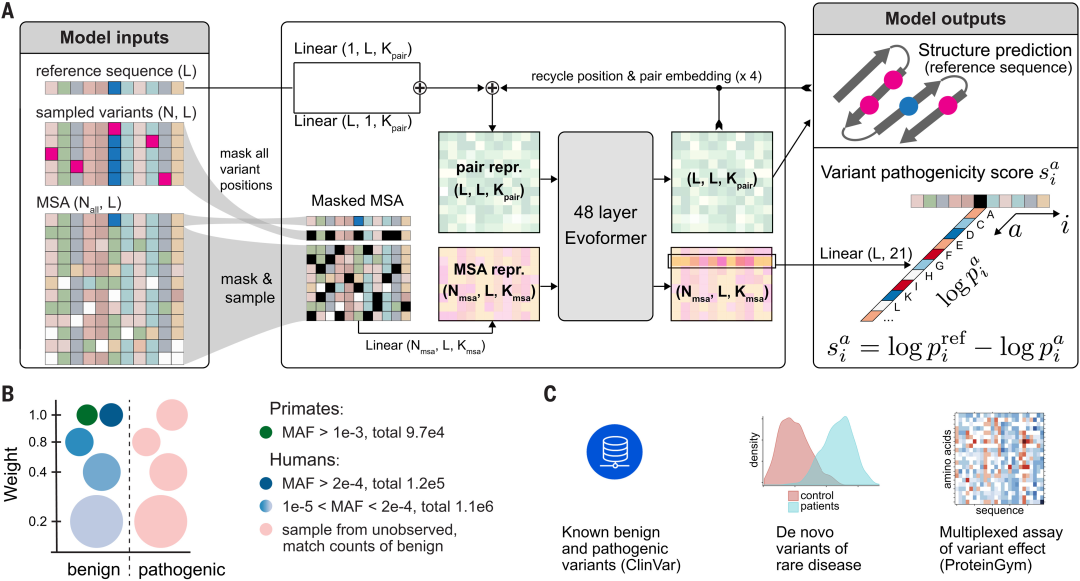
这项最新研究开发了 AlphaMissense ,并利用了多个方面的进展:(i) 无监督蛋白质语言模型,以学习序列上下文条件下的氨基酸分布;(ii) 使用源自 AlphaFold 的系统纳入结构背景;(iii) 对来自种群频率数据的弱标签进行微调,避免来自人工编辑注释的偏差。
在临床注释、新生疾病变异和实验分析基准中,AlphaMissense 实现了最先进的错义致病性预测,而无需特别在这些数据上进行训练。作为社区资源,该研究为人类蛋白质组中所有可能的单氨基酸取代提供了一个预测数据库。研究在 ClinVar 数据集上使用精确度 90% 的截止值,将32%的错义变异分类为可能致病的,57%的错义变异分类为可能良性的,从而为大多数人类错义变异提供了可靠的预测。
研究展示了如何利用这些资源来加速多个领域的研究。分子生物学家可以使用该数据库作为起点,设计和解释探测人类蛋白质组中饱和氨基酸取代的实验。遗传学家可以将基因水平的 AlphaMissense 预测与基于群体队列的方法相结合,以量化基因的功能意义,特别是对基于队列的方法缺乏统计能力的较短的人类基因。临床医生在为罕见疾病诊断优先考虑新生变异时,可以从可靠分类的致病变异的覆盖范围增加中受益,并且 AlphaMissense 预测可以为使用罕见的、可能有害的变异注释的复杂性状遗传学研究提供信息。
AlphaMissense 预测可能阐明变异对蛋白质功能的分子效应,有助于鉴定致病性错义突变和以前未知的致病基因,并提高罕见遗传疾病的诊断率。AlphaMissense 还将促进从结构预测模型中进一步开发专门的蛋白质变异效应预测因子。
图1. AlphaMissense 致病性预测。AlphaMissense将错义变异作为输入并预测其致病性。研究根据人类和灵长类动物变异种群频率数据对 AlphaFold 进行了微调,并校准了已知疾病变异的置信度。AlphaMissense 预测错义变异致病性的可能性,并将其分类为可能良性、可能致病性或不确定。研究为所有可能的人类错义变异提供预测,作为社区资源。
大模型与生物医学:
AI + Science第二季读书会启动
详情请见:
大模型与生物医学:AI + Science第二季读书会启动
推荐阅读