Nature速递:AlphaFold 3 预测所有生命分子的结构和相互作用
导语


网络架构和训练
网络架构和训练

不同复合物类型的准确度
不同复合物类型的准确度

预测的置信度与准确性相一致
预测的置信度与准确性相一致

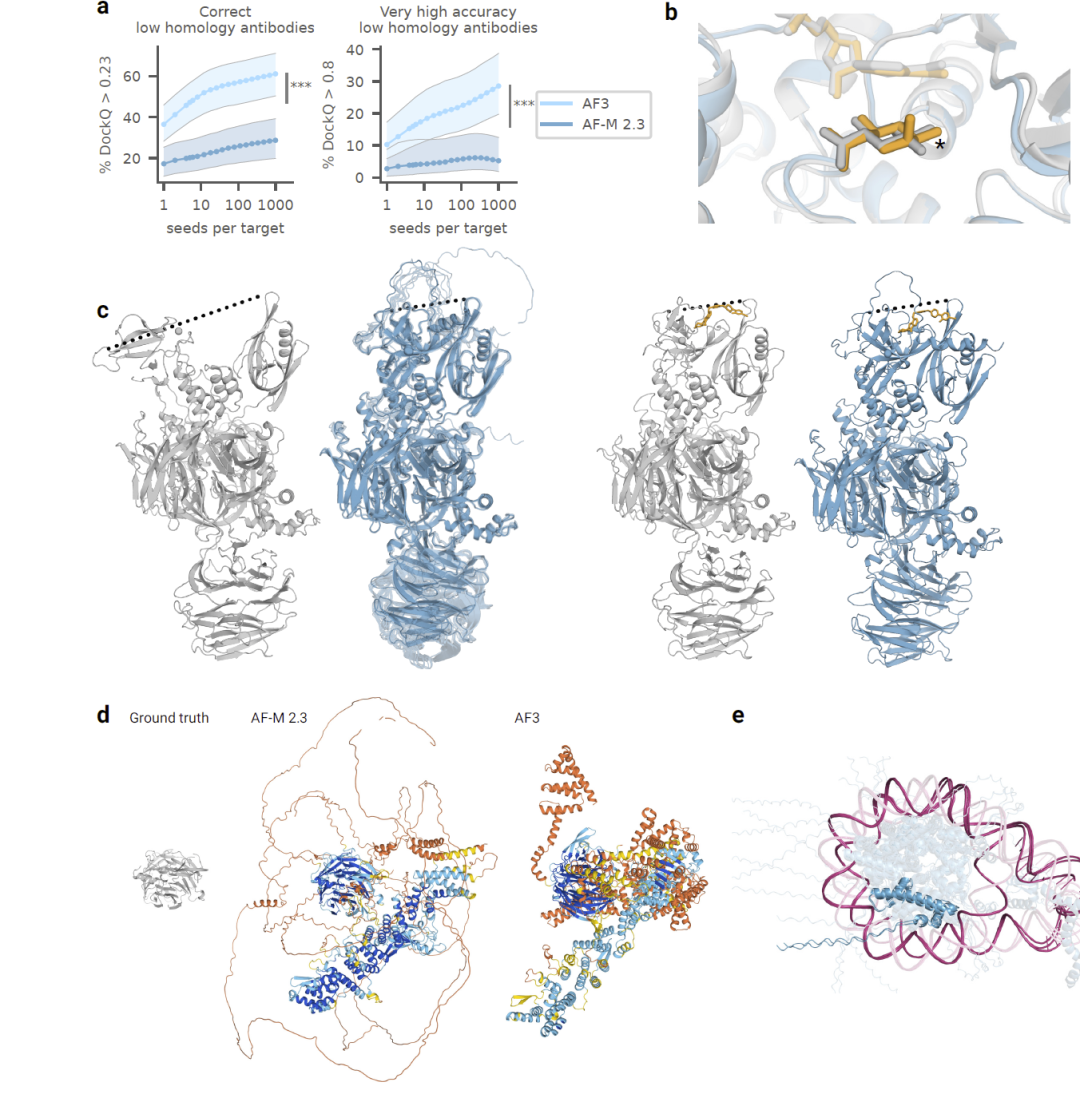
模型局限性
模型局限性
结论
结论
1. Abramson, J., Adler, J., Dunger, J. et al. Accurate structure prediction of biomolecular interactions with AlphaFold 3. Nature (2024). https://doi.org/10.1038/s41586-024-07487-w
2. https://blog.google/technology/ai/google-deepmind-isomorphic-alphafold-3-ai-model/
3. https://www.isomorphiclabs.com/articles/alphafold-3-predicts-the-structure-and-interactions-of-all-of-lifes-molecules
4. https://www.isomorphiclabs.com/articles/rational-drug-design-with-alphafold-3
大模型与生物医学:
AI + Science第二季读书会
详情请见:
微信扫一扫,分享到朋友圈











