人工智能终结了蛋白质研究吗?一文读懂蛋白质折叠的前世今生
来源:Fran Pulido
导语
2020 年 12 月,由于新冠疫情限制了大家的活动,许多人无法亲自参加会议,数百名计算科学家聚集在屏幕前,共同见证科学新时代的到来。
他们聚集在一起是为了参加一个会议,一个持续近三十年的友谊赛。他们其中有些人亲身参与过这项赛事,并通过这项比赛同聚一堂,痴迷于探讨同一个问题——这便是蛋白质折叠问题。简单来说就是:我们能否根据最基本的信息——也就是蛋白质分子的一维分子编码——准确预测出其三维结构?蛋白质让人类细胞和身体保持活力和正常运转。由于蛋白质的结构决定了它的行为,因此成功解决这个问题将对我们了解疾病、研发新药和理解生命体的运作方式产生深远影响。
在每两年举行一次的会议上,科学家们将用最新的蛋白质折叠工具做测试。但解决方案似乎总是遥不可及。有些人穷极一生都在力求逐步提高预测的准确性,因此这项赛事只能说是蹒跚前行,参赛的研究者们没有理由认为2020年会有所不同。
但他们想错了。

比赛当周,蛋白质科学界的新秀John Jumper展示了谷歌 DeepMind在伦敦的人工智能分部推出的最新人工智能工具:AlphaFold2。他通过Zoom会议分享的相关数据显示,AlphaFold2的三维蛋白质结构预测模型准确率超过90%,比最接近的竞争对手高出5倍。
一瞬间,蛋白质折叠问题的解决方案从遥不可及变为了唾手可得。人工智能轻松地蹚过了人类智慧举步维艰的泥潭,这震撼了整个生物学界。出席会议的哥伦比亚大学数学基因组学项目的系统生物学家Mohammed AlQuraishi表示:“我感到非常震惊。很多人都拒绝接受这个现实。”
但在总结陈词时,会议组织者John Moult打消了所有疑虑。他几乎斩钉截铁地表示:AlphaFold2已经“基本解决”了蛋白质折叠问题,并永远改变了蛋白质科学。他穿着黑色高领毛衣,坐在家中办公室的书架前,用Zoom分享着自己的幻灯片,“这不是结束,而是开始,”他这样说道,语气既令人兴奋又令人担忧。
当谷歌的公关部门向全世界发布这一消息时,媒体为之疯狂。AlphaFold2“将会改变一切”的说法占据了各大新闻的头版头条。那些毕生致力于研究单个蛋白质结构的蛋白质生物学家担心自己会丢掉饭碗。还有人声称,AlphaFold2将彻底改变药物研发,因为生物学家可以快速了解蛋白质的结构,从而更高效地研发出以蛋白质为靶点的新药。其他人则反驳说,这些结果大多是炒作,不会带来实质性改变。
Moult自己也几乎无法理解这件事本身意味着什么。他在会议最后问出了大家都想问的问题:“接下来怎么办?”
这个问题已经是三年半以前的事了,现在我们终于能够尝试回答他的问题了。
不可否认,AlphaFold2的确改变了生物学家研究蛋白质的方式。然而,虽然AlphaFold2是一个强大的预测工具,但它并不是一个全知全能的机器。它非常巧妙地解决了蛋白质折叠问题的一部分,但并不是科学家所想的那样。它并没有取代生物实验,而是强调了生物实验的必要性。
AlphaFold2最大的影响可能是让生物学家关注人工智能的力量。它已经启发了新的算法,包括设计在自然界中不存在的新蛋白质的算法。此外,它还催生了新型生物技术公司,引领了新的科学实践方式。2024年5月,其继任者AlphaFold3发布,它能够模拟蛋白质和DNA或RNA等其他分子的结构与相互作用,标志着生物领域的预测模型进入了新的发展阶段。
AlQuraishi表示:“这是迄今为止科学领域最重要的‘机器学习’故事。”
然而,生物科学领域仍有许多人工智能未能跨越的鸿沟。这些工具无法模拟蛋白质如何随时间发生变化,也无法根据蛋白质存在的环境(即细胞内)对其进行建模。在劳伦斯伯克利国家实验室开发生物分子结构建模算法的结构生物学家Paul Adams表示:“AlphaFold似乎改变了一切,又好像什么都没有改变。”
本文便是有关来自谷歌DeepMind的Jumper团队是如何变革蛋白质科学及其如何影响人工智能在生物学领域未来应用的故事。
1. 前夜
1. 前夜
(1)物理基础
一张折纸在以特定方式折叠之前,只不过是压制的木浆,而在折叠之后,它才能焕然一新。几经精妙的卷曲与翻折,它就变成了一个纸制的算命工具,可以用来预测你的未来。而只需改变几个折叠步骤,同一张纸就可以变成一只展翅飞翔的仙鹤,给人带来好运。
同样,一长串氨基酸分子在自发折叠成其固有形状(生物学家称之为结构)之前没有任何功能。蛋白质的结构决定了它如何与其他分子结合或相互作用,因此也决定了它在细胞中的作用。
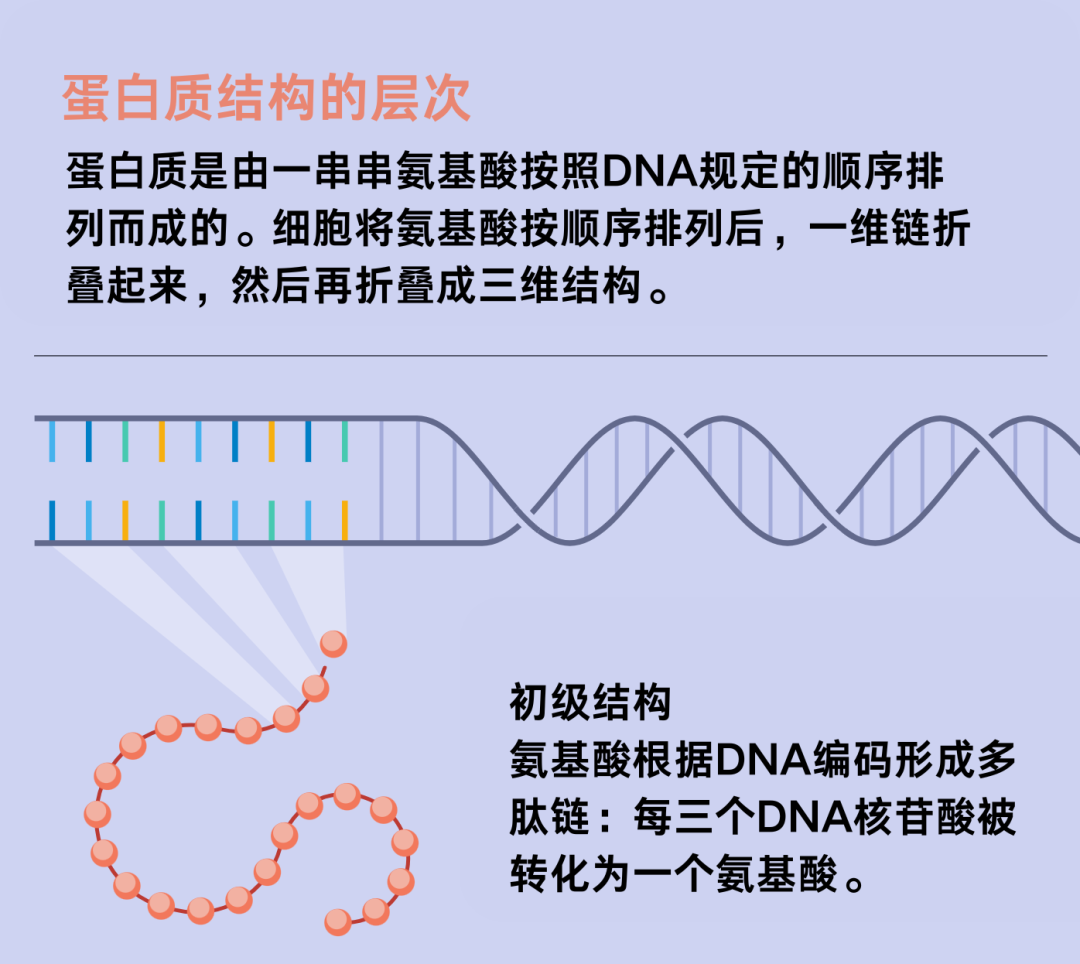
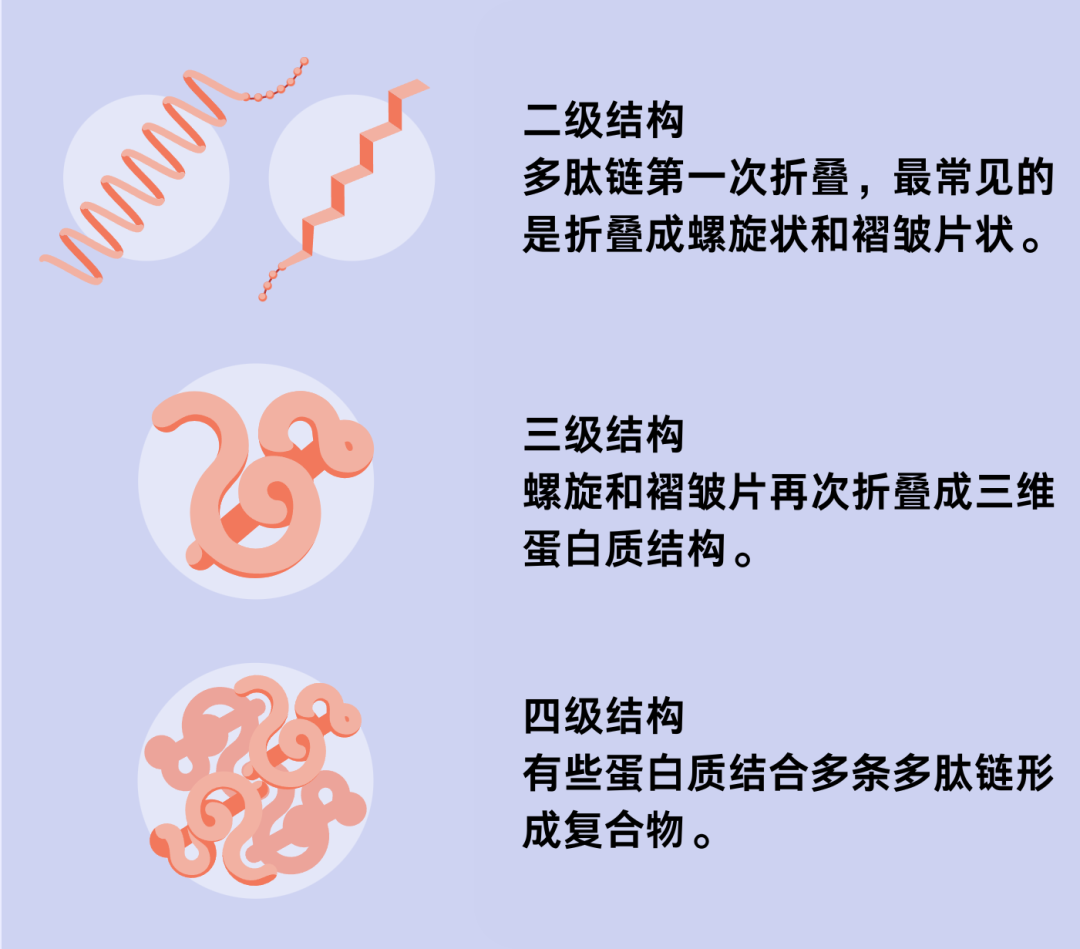
图源:Mark Belan,翻译:vicky,制图:存源
地球上有几亿种已知的蛋白质,还有更多未知的蛋白质。它们无所不能:血红蛋白和肌红蛋白将氧气运送到肌肉和身体各处。角蛋白构成头发、指甲和皮肤的外层结构。胰岛素使葡萄糖进入细胞,转化为能量。蛋白质的形态似乎变幻莫测,以满足实际生活中各种各样的需求。
“从原子到生态系统,(蛋白质结构)像是一种通用语言,是一切的起源,”AlQuraishi说道。
细胞通过菊花链的方式将氨基酸这种小分子连接成长长的多肽串,从而生成蛋白质。它所选择的氨基酸取决于DNA提供给它的一组基本指令。生成后不到一秒,多肽串就会开始精确地弯曲、扣合、折叠成蛋白质的最终三维结构。一旦装配完成,它们便会立刻开始履行自己的生物职能。
如果蛋白质不能很好地完成这一折叠过程,那么人体将会经受一系列危及性命的打击。错误折叠或结构解开的蛋白质会导致中毒和细胞死亡。许多疾病和失调都是由错误折叠的蛋白质引起的,如镰状细胞贫血。此类蛋白质还会聚集成块,这也是阿尔茨海默病和帕金森病等神经退行性疾病的特征。
然而,没有人真正了解蛋白质是如何折叠的。这些简单分子链中的序列信息又是如何编码蛋白质的复杂结构呢?约翰霍普金斯大学生物物理学名誉教授George Rose表示“这是我们能提出的最深奥的问题。”

早在20世纪30年代,科学家们就开始探究这个问题。但真正取得突破要等到20世纪50年代中期,当时一位名叫Christian Anfinsen的生物化学家将蛋白质加入化学溶液中,这一做法要么会导致蛋白质的键断裂,使其结构展开;要么会导致蛋白质发生错误折叠。通过观察,他发现,展开或折叠错误的蛋白质可以自发地重新折叠成正确的结构。这一发现证明了蛋白质的三维结构是由内部编码(由氨基酸串的编码)决定的,Christian Anfinsen也凭此获得了诺贝尔奖。
基于这一发现,Anfinsen提出了一个假设:应该有一种方法可以根据氨基酸序列预测蛋白质的结构——这就是后来广为人知的蛋白质折叠问题。
一旦多肽链组装完成,它们就能在千分之一秒内折叠成正确的结构,速度之快令分子生物学家Cyrus Levinthal咋舌。在1969年发表的论文《如何优雅地折叠》(How to Fold Graciously)中,Levinthal计算出,如果一个蛋白质要尝试每一种可能的折叠方案,那么它的组装时间将长得无法想象。他推测,显然是有某种更为直接地方式引导了蛋白质的正确折叠。
随着时间的推移,蛋白质折叠问题又衍生出新的问题,主要有三个:能否根据氨基酸序列预测蛋白质的结构?折叠编码是什么?折叠机制是什么?
20 世纪 60 年代初,当第一批通过实验决定的蛋白质结构问世时,这些问题开始在科学家的心中生根发芽。剑桥大学的两位生物学家Max Perutz和John Kendrew将蛋白质培育成晶体,用X射线轰击它们,并测量射线如何弯曲——这种技术被称为X射线晶体学(X-ray crystallography)。通过这种方法,他们确定了血红蛋白和肌红蛋白的三维结构。这项实验耗时二十多年,为两人赢得了诺贝尔奖。

从那时起,无数研究人员不懈努力,试图了解不同蛋白质的结构,及其底层的机制。格拉斯哥大学的结构生物学家Helen Walden表示:“想弄清楚事物的外观是人类的本能,因为这样我们便能了解其功能。”还有一些科学家尝试从化学或物理的角度解决问题。他们日复一日地在实验室里辛勤工作,最终成功重建了蛋白质的结构。计算生物学家通过模型和模拟寻找线索,他们用不同的算法规则组合对模型和模拟进行编程并不断改进更新。
随着越来越多的蛋白质结构被发现,蛋白质科学界需要一种方法来组织和共享这些信息。1971年,蛋白质数据库(Protein Data Bank),作为一个存储蛋白质结构的档案库,应运而生。该数据库可免费使用,对于所有想要通过了解蛋白质结构来探究生物问题的研究者来说,都是一个可靠的工具。
蛋白质数据库建立之初,只保存了7种蛋白质的结构。将近50年后,当谷歌DeepMind利用它来训练AlphaFold2时,它已记录了超过14万种蛋白质结构——每个结构都是由结构生物学家费尽周折解码出来的。
(2)实验派的苦恼
从20世纪70年代中期起,牛津大学的生物物理学家Janet Thornton每隔几个月就会收到一个邮包。里面是一卷12英寸的磁带,包含了存入蛋白质数据库的新蛋白质结构数据。她会迫不及待地撕开包裹,想要立刻开始分析新发现的蛋白质结构。她收到的第一盘磁带上只包含20种蛋白质结构的数据。

磁带记录的每一个蛋白质都是多年心血的结晶。通常情况下,一名博士生会在读研的四年间或花费更长时间研究单个蛋白质的结晶,从中收集数据或解读数据,从而厘清其折叠结构。
牛津大学生物物理系当时是世界X射线晶体学中心之一。1965年,蛋白质晶体学的先驱之一David Phillips在牛津大学首次测定了溶菌酶的结构,这种酶会被免疫系统用来对抗细菌。牛津大学的生物物理学家利用X射线晶体学绘制了蛋白质的电子密度图;电子聚集的区域很可能含有一个原子。Thornton和她的同事们将这些电子密度图打印到塑料板上,然后将它们叠放在一起,从而绘制出蛋白质的形态“轮廓图”。

随后,他们将轮廓图转换成了物理模型。他们先将塑料平面图放入理查兹盒中——以牛津大学生物物理学家弗雷德里克·理查兹(Frederic Richards)命名的装置,他于1968年发明了这种装置。理查兹盒内设有一面倾斜的镜子,能将轮廓图反射到一个工作区内,这让科学家们能看到每个原子相对于其他原子的准确位置。然后,他们用小球和棍子搭建出了一个物理模型。
这种方法既繁琐又有局限性。1971年,Louise Johnson着手为磷酸化酶建模,包含842个氨基酸的磷酸化酶是当时学界研究过的最大的蛋白质。为了成功建立模型,Johnson不得不爬梯子进入牛津大学专门为她的项目建造的两层楼高的理查兹盒中。她日后成为了知名晶体学家。
模型完成后,科学家们用尺子测量了原子间的距离,从而得出蛋白质结构的各个坐标。Thornton表示:“这种方法太过时了。”然后,他们将坐标输入计算机。她说,电脑上看起来就像一片茂密的森林,原子们杂乱无章地挤在一起。只有戴上3D眼镜观察结构时,才能看清蛋白质的拓扑结构。
“这个过程太折磨人了,能坚持到底已经非常了不起了,”Thornton感慨道。
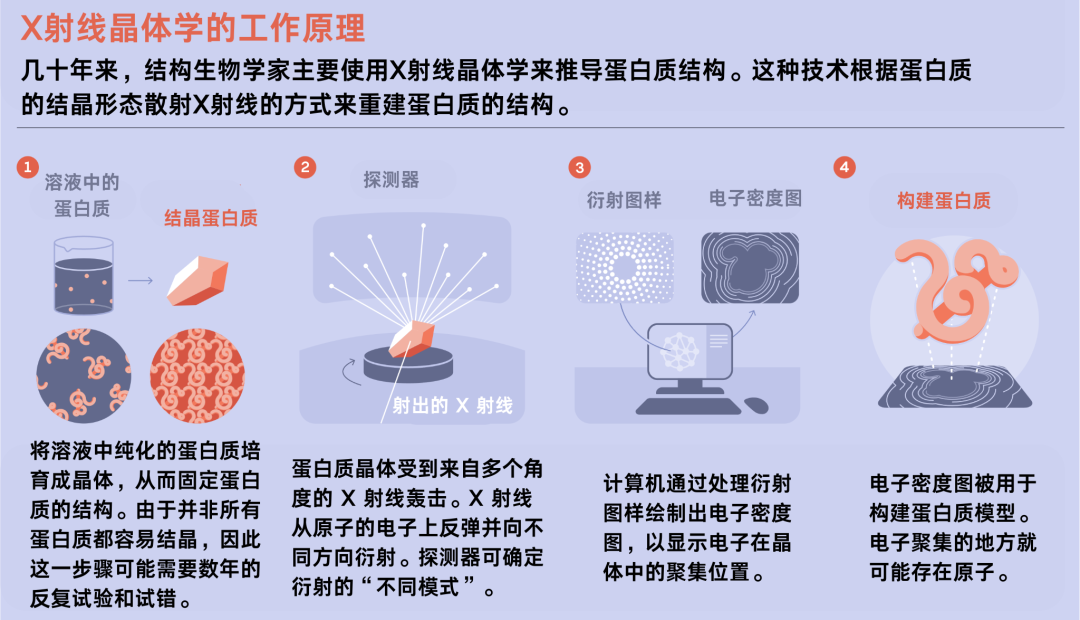
X射线晶体学的工作原理。
年复一年,他们终于成功了。一旦研究人员确认了自己所复构的蛋白质结构,他们就会将其提交给蛋白质数据库。到1984年,已有152个蛋白质的结构被存入了数据库。1992年,这一数字攀升至747个。
当实验学家们还在苦苦研究物理模型时,另一学派的蛋白质生物学家——计算科学家们——却采取了不同的方法。但是,当他们考量Anfinsen所提出的假设,即根据氨基酸序列预测蛋白质结构时,却显得有点过于自信了。
(3)制定规则
20世纪60年代初,John Moult还是一名大学生时,就计划成为一名物理学家。后来,他了解到了蛋白质折叠问题。他说:“有人来给我们讲了一堂课,说生物学问题太重要了,不能留给生物学家来解决。我当时很傲慢,所以当真了。”被生物学迷住的他将职业生涯转向了另一个方向。
毕业后,Moult进入了蛋白质晶体学领域。他解码了几种蛋白质的结构,包括β-内酰胺酶,这是一种能够破坏青霉素的细菌酶。1970年,他在牛津大学获得分子生物物理学博士学位。但当开启博士后研究后,他厌倦了实验派的方法,并开始转向逐步发展壮大的计算派。计算生物学家与实验学家不同,他们编写计算机算法,试图证明Anfinsen的假设,即他们可以向一个程序输入一串氨基酸,从而生成正确的蛋白质结构。
从生物实验到计算的转变并非坦途。Moult已经习惯了以慢工出细活地方式解析单一蛋白质的结构。但在计算生物学领域,每隔一段时间就有论文声称蛋白质折叠问题和相关子问题已经得到了解决。
Moult对此表示怀疑。他说:“在这一领域发表的论文不像我以前所在领域那样严谨。这并不是说计算生物学领域都是骗子,而是因为此类计算工作都是在虚拟世界中完成的。”
在虚拟世界中,当自然界的规则不起作用时,计算学家们就会制定自己的规则。他们会设计算法,让原子以某种方式粘在一起,或者让蛋白质总是向右或向左折叠。随着时间的推移,这些模型变得越来越脱离现实。Moult认为,在一个你可以完全控制的世界里,人很难保持严谨。
不过,他还是认为两种方法都有可取之处。实验派工作精细,但速度缓慢;计算派进展迅速,但却远离生物物理现实,以至于经常出错。
他想,一定有办法把两种方法的优点结合起来。
(4)跺脚以示反对
20世纪90年代初,Moult和他的同事Krzysztof Fidelis想出了一个办法来规范计算生物学领域的混乱局面。他们启动了一个名为“结构预测关键评估”(Critical Assessment of Structure Prediction,简称 CASP)的社区科学实验。
这个想法很简单。作为CASP的发起人,Moult和Fidelis会公布一份蛋白质氨基酸序列列表,上面列举了近期已被解析的蛋白质结构,相关数据由相应的实验人员提供,但结果尚未发布。然后,世界各地的计算研究小组将使出浑身解数来预测蛋白质的结构。一个独立的科学家小组将通过比较他们的答案和实验证实的结构来评估这些预测模型。
这个想法被付诸实践。于是,CASP很快就成为以计算方法解决蛋白质折叠问题的试验场。那时还没有人工智能,计算方法主要是模拟分子的物理学原理。这是科学家们公开与同行同场竞技,验证自己观点的机会。Thornton表示:“CASP本意并不是为了比赛,但实际上已经成为一项赛事。”
CASP每两年举办一次,科学家们会在阿西洛马会议中心齐聚一堂,这是加利福尼亚州蒙特雷附近一座古老的小教堂,曾经是卫理公会教徒的隐居地。与会期间,组织者会宣布竞赛结果,计算学家们会发表演讲,介绍自己所用的方法。Moult鼓励与会者如果不赞同陈述的内容,可以在木地板上跺几脚以示反对。
他说:“刚开始的时候,跺脚声确实是此起彼伏。”
曾师从Thornton的伦敦大学学院生物信息学教授David Jones回忆说:“跺脚声仿佛鼓声一样。”如果演讲者所分享的细节太冗长,生物学家们会跺脚;如果陈述内容夸大其词,他们也会跺脚;如果演讲者的观点重复或过于杂乱,他们还会跺脚。Jones表示:“跺脚声并不讨厌,”因为这都是善意的声音。

不管跺脚是出于什么原因,当跺脚声在演讲者耳边响起时,都会让人感到尴尬。Jones说:“感谢老天爷,我演讲时没人跺过脚。有一年,他和同事们提出了一种名为“穿线法(threading)”的计算方法,即通过已知的蛋白质结构编织氨基酸序列,寻找契合点。他们的表现还可以。“我们当时很高兴。……但之后就开始走下坡路了,”Jones笑着回忆道.“真的,挺很意思的。”
帕多瓦大学生物信息学教授Silvio Tosatto表示,当时学界兴致盎然。“大家觉得如果自己有能够正确预测蛋白质结构的算法,就可以成为百万富翁。还有一些人认为诺贝尔奖已经近在咫尺了。”
当然,早年这些美梦都没成真。当被问及在此期间,CASP收到的分享报告质量如何时,Moult停顿了一下,说道:“可以用参差不齐(Random)来形容。”
有些方法的效果比预期的要好,比如“同源建模(homology modeling)”,它通过比较已知蛋白质的结构来推导未知蛋白质的结构。还有些方法则一败涂地。Moult说,大多数模型的预测结果都是“扭曲变形的”。
荷兰癌症研究所和乌特勒支大学的结构生物学家Anastassis Perrakis开玩笑说:“我很喜欢看到他们失败。”他曾将实验验证的蛋白质结构提供给CASP组织者作为比赛参考。“这不是较劲,我们只是喜欢就科学问题捉弄彼此。”
不过在这一过程中,也有一些人崭露头角。1996年,第二届CASP结束后,一位名叫David Baker的年轻人邀请Jones共乘一辆出租车前往机场。他听过Jones的演讲,正在研究自己的计算模型。虽然他还没准备好参加这次的CASP,但他想和Jones聊聊。于是,Jones在出租车上倾听了他的想法,但没想过会再见到他。
等到1998年,这位年轻人凭借自己的算法Rosetta惊艳全场。Jones说,他当时可谓是“一马当先”。

Rosetta这样的算法可以模拟氨基酸分子的原子间相互作用,从而预测它们将如何折叠。它们“表明你确实可以预测蛋白质结构。但它还不够好,也不够准确,因此实用价值有限,”Baker评论道。
2008年,计算机的表现依然不如人类。Baker当时正在华盛顿大学管理自己的实验室,他创建了一款名为Foldit的免费在线游戏,玩家可以将一串串氨基酸折叠成蛋白质结构。他的团队在发表于《自然》期刊的一篇论文中报告说,人类玩家在蛋白质建模方面的表现优于Rosetta。
但人类的领先优势不会持续太久。2010年代初,科学家们在一个被称为“协同进化(co-evolution)”的概念上取得了重大突破,这推动了这一领域的发展,并为日后人工智能的发展打下了重要基石。这个概念其实已经存在了几十年,简单明了的说就是:通过比较成百上千个蛋白质中密切相关的氨基酸序列,科学家们可以识别出发生变异的氨基酸。更重要的是,可以确定它们是否与其他氨基酸同步发生变异。如果两个氨基酸同时发生了变化,那么它们之间很可能存在某种联系。伯克利实验室的结构生物学Adams表示:“我们可以说,这两个氨基酸也许在空间上很靠近。”
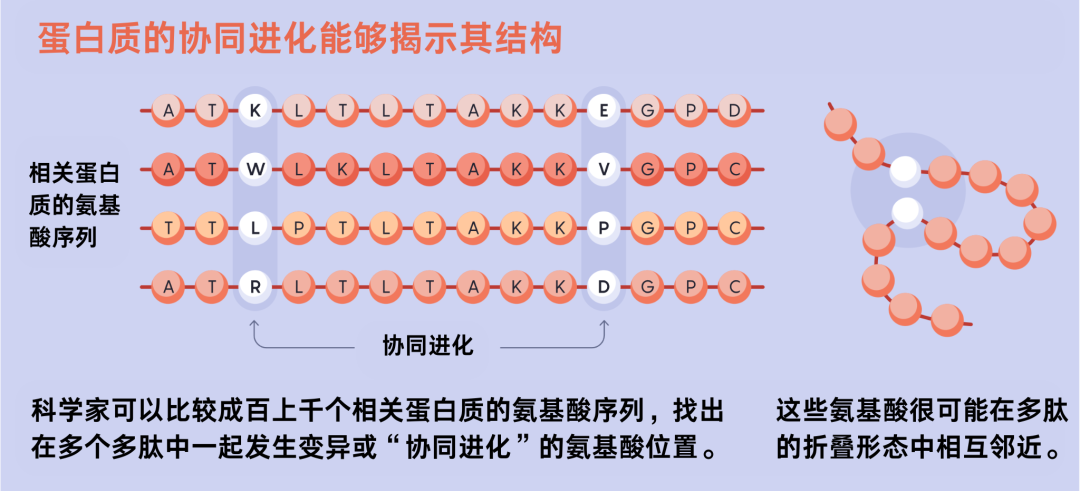
蛋白质的协同进化能够揭示其结构。图源:Mark Belan
但直到2010年代初,这种预测哪些氨基酸会发生接触的方法令人失望,准确率徘徊在20% 到24%之间。随后,科学家们注意到,他们的统计方法存在误差,也就是说一些看似彼此接触的氨基酸实际上并没有任何接触。后来,Moult了解到,几十年来,统计学家一直对这种误差的存在心知肚明。他说,回首往事,你会想“我当初怎么这么笨?”
随后,计算生物学家清理了统计工具。到了2016年,接触预测的准确率已经飙升至47%。两年后,准确率达到了70%。Baker的算法之所以能取得成功还要归功于以下原因:2014年,Rosetta生成了两个非常精确的蛋白质结构,以至于CASP评审员认为Baker可能已经解决了蛋白质折叠问题。
Adams表示,协同进化的洞见“非常奇妙”。在不使用机器学习的情况下,协同进化是“真正推动该领域发展的重大事件之一”。
然而,这项突破并没能带领该领域走得太远。协同进化需要大量相似的蛋白质进行比较,而实验人员解析蛋白质结构的速度不足以满足计算人员的需求。
谈及这一切时,Moult引用了进化生物学中的一个术语:岁月在间断平衡(punctuated equilibrium)中流逝。有时,人们感觉好像十亿年都没有进化出好的想法,但紧接着便有可能发生令人兴奋的进展。
2. 问世
2. 问世
(1)仓促上阵
2016年,David Jones在《自然》期刊上发表的一篇新论文中瞥见了未来的景象。来自谷歌DeepMind(一个位于伦敦的人工智能团队)的研究人员详细介绍了他们的算法是如何利用一种被称为深度学习的方法,在围棋这项古老的棋盘游戏中击败人类冠军的。
Jones大吃一惊。“大事即将发生,我真的要了解深度学习了,”他回忆道。
深度学习是一种受人类大脑启发的人工智能技术。在你的大脑中,分子信息通过由神经元这种脑细胞构成的互通互联网络进行传输。神经元向外延伸的突起叫做树突,可以捕捉邻近神经元派出的分子,这些分子会告诉接收神经元,是否需要发射和传播信号。
布朗大学计算机科学系的教授Michael Littman说:“如果该神经元接受到了足够多的活动分子,那么该神经元就会被激活,从而发出信号。这将导致另一波分子被释放到下一个神经元。”
20世纪50年代,计算机科学家意识到,他们可以将电子比特连接起来,创建“神经网络”。神经网络中的每个单元都是一个节点,研究人员将其比喻为神经元:一个神经元接收来自其他神经元的信息,然后计算是否向下一个神经元发射信息。在这样的神经网络中,信息通过多层神经元传播,产生特定的结果,比如识别图像中的狗等。

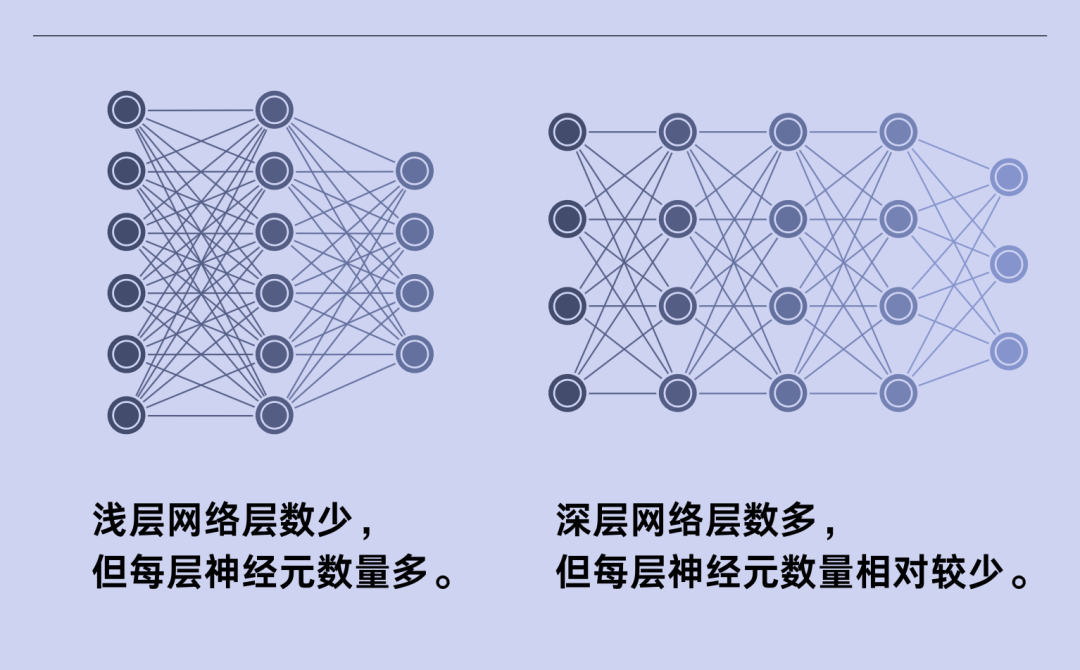
神经网络的工作原理。图源:Mark Belan
神经元的层数越多,能进行的计算越复杂。但早期的神经网络一般只有两层。到了20世纪90年代,神经元层数增加到了三层,并在接下来20年间都未增加。Littman说:“我们不知道如何可靠地创建比这更深层的神经网络。”
自20世纪90年代以来,包括Jones和Moult在内的结构生物学家一直尝试将神经科学网络运用到蛋白质科学中,但浅层网络和稀疏数据的局限性让他们止步不前。随后时间来到20世纪10年代初,计算机科学家们学会了如何更好地构建神经网络,以便对更多层神经元进行稳定的训练。神经网络的层数也逐渐从20层、50层、100层发展到数千层。Littman表示:“为了区别于我们在90年代的做法,人们开始称之为‘深度学习’。要说机器学习领域的研究者们擅长什么,那可以说是创造一些酷炫的名词了。”
深度学习改变了人工智能,使算法能够出色地识别照片或声音中的特征,并在游戏中击败了人类。
2016年3月,当DeepMind联合创始人Demis Hassabis在首尔见证其旗下的人工智能系统AlphaGo在历史悠久的围棋比赛中击败人类世界冠军时,他回想起了自己在大学时玩Foldit的情景。他想知道,如果DeepMind的研究人员能写出模仿围棋大师直觉的算法,难道他们不能写出模仿Foldit游戏玩家直觉的算法吗?这样的算法虽然不懂生物学,却知道如何折叠蛋白质。

芝加哥丰田工业大学的教授许锦波(Jinbo Xu)也认识到了利用深度学习解决蛋白质折叠问题的潜力。他受到了神经网络在图像识别方面所取得的成绩的启发。当时,计算机科学家已经在卷积网络方面取得了巨大成功,卷积网络利用深度学习算法将图像分割成若干块,并识别它们之间的模式。许教授将这一技术引入了蛋白质折叠领域。他使用一种称为矩阵的数学对象来表示哪些氨基酸在空间中彼此邻近,然后将数据作为图像输入卷积网络。该算法通过寻找这些图像之间的模式来预测组成蛋白质的原子的三维坐标。
2016年,他在arxiv.org上发布了这项的预印*(后来发表在PLOS Computational Biology上),向人们展示了“深度学习的潜在应用”。Moult表示:“这在当时对蛋白质折叠领域影响颇深。”
Wang, Sheng, et al. “Accurate de novo prediction of protein contact map by ultra-deep learning model.” PLoS computational biology 13.1 (2017): e1005324.
不久之后,蛋白质结构研究小组开始涉足深度学习。AlQuraishi和他的团队率先开发出了一种方法,可以完全通过神经网络直接预测蛋白质结构,也就是所谓的“端到端”(end-to-end)方法,只是效果并不理想。其他人则是各显神通,努力想要跟上这波科研的新浪潮。
Jones说:“我当时并不清楚自己想用深度学习做什么,但我意识到我需要了解和运用深度学习。”
在他已经开始撰写资助申请,自寻出路时,他收到了谷歌DeepMind的一封电子邮件。他们向Jones询问了CASP竞赛的情况,并表示愿意提供帮助。“我以为他们的意思是:我们可以提供强大的计算机能力,”琼斯说道。
双方见面之后,Jones发现谷歌的野心显然不止于此。但要实现自己的雄心壮志,这家科技巨头公司需要更多的科学人才。
(2)新秀入场
2016年,Jones开始担任谷歌DeepMind的顾问,负责一个后来被称为AlphaFold的项目;与此同时,John Jumper正在芝加哥大学完成他的理论化学博士学位。
少年时期,Jumper自学了计算机编程。他在物理方面也很有天赋。因此,到了选择大学专业时,尽管他的父母都是工程师且担心他找不到工作,但他还是决定学习数学和物理。
“我一直以为自己会成为一名研究‘宇宙法则’的物理学家,”Jumper说。“我一直热爱探索宇宙真理的这一理念。”

在范德比尔特大学读本科时,他与费米国家加速器实验室的研究人员合作,研究夸克(quarks)这种亚原子粒子的奇特属性。一天,当他和研究人员坐在一起吃午饭时,他得知了一个坏消息。“那么,我们正在进行的这项实验什么时候才能启动呢?”Jumper回忆自己曾这样问他们。其中一位教授说,他可能退休前等不到了。另一位年纪稍长的教授说,他可能都活不到那一天。
“我希望自己做科研耗费的时间比两位教授说的短一点,”Jumper说。在完成本科学业后,他参与了一个理论凝聚态物质物理学的博士课程——但很快就辍学了。他在D.E. Shaw研究公司找到了一份工作,这是一家纽约的公司,当时正在进行蛋白质基本模拟的研究。通过了解蛋白质如何运动和变化,他们希望能更好地理解各种疾病的机制,例如肺癌。
这是Jumper第一次意识到自己工作的潜在重要性。“这关乎健康和延长人们的寿命,”他说。在接下来的三年里,Jumper在公司的超级计算机上对蛋白质的运动进行了建模,这些超级计算机是专门为更快地模拟分子而建造的。“有时候,我在周二一天做的模拟比我整个博士期间要做的还多,”他说。
2011年,他决定再次尝试读研,这次是在芝加哥大学学习理论化学。他仍然对蛋白质结构和运动感兴趣。但他对学术界的缓慢进展感到沮丧。“我再也无法使用我在D.E. Shaw公司用过的那种定制计算机硬件了,”Jumper说。他想知道是否可以使用人工智能(当时被称为统计物理)来实现快速模拟,而这通常需要先进的机器。他开始涉足机器学习和神经网络。
在此期间,他也开始思考蛋白质折叠问题。他觉得这个问题应该可以用蛋白质数据库中的训练数据来解决——到2012年,该数据库已包含超过76,000种蛋白质结构。
Jumper说:“我当时相信数据量已经足够多了,但是想法还不够成熟。”
2017年,Jumper听说谷歌DeepMind开始涉足蛋白质结构预测。他刚刚完成博士学位,读博期间恰好主攻利用机器学习来模拟蛋白质折叠和动态。于是,他申请了谷歌的研究科学家职位。
“这个项目当时处于保密阶段,”他说。如果他在面试中提到蛋白质折叠,DeepMind团队就会转移话题。“您转移话题太多次的话,只会让我更加确定贵公司在做什么。”Jumper说。

谷歌 DeepMind 总部位于伦敦。图源:Buildington
2017年10月,他来到了DeepMind位于伦敦的办公室。在顾问Jones的帮助下,团队已经开始深入开发AlphaFold。“那会儿真的太有意思了,我们不断提出各种想法,”Jones说。“最终会有一个比较好的核心想法脱颖而出,然后他们便开始全力推进。”
为了训练算法,DeepMind团队使用了超过140,000个来自蛋白质数据库的蛋白质结构。他们将这些信息输入到一个卷积网络中,但并没有对AI架构本身进行太多改动。“这是‘标准的机器学习’,”Jumper说道。
到了2018年春季,AlphaFold准备参加CASP,与“正统的”蛋白质科学家们同台竞技。“有点像一级方程式赛车,”Jones回忆道。“你认为自己建造了最好的赛车,但你不知道其他车队的实力如何。”由于风险很高,DeepMind团队讨论是否应匿名参赛,因为他们不想被当众羞辱。
Jones说:“没有人想失败。”在学术界,失败是工作的一部分;你失败了,只能继续前行,因为你别无选择。“但如果是一家价值数十亿美元的科技公司,那么失败的尝试显然会很让公司看着狼狈。”
他们最终决定以谷歌DeepMind的名义提交参赛成果。在12月会议开始前几个月,Jones收到了CASP组织方的回复,他们建议DeepMind团队线下参加会议,因为AlphaFold表现非常出色。
虽然他们在预测蛋白质结构方面的准确性大约是第二名的2.5倍,但并非绝对碾压。不过,他们的凯旋依旧给人留下了深刻印象。“显然,有趣的事情正在上演,”Moult说道。
(3)重启算法
这次胜利对DeepMind团队来说本应是一针强心剂,但他们知道自己离真的解决蛋白质折叠问题还相距甚远。几个月前,Hassabis将团队成员召集在一起。“我们的目标是否是真正解决这个问题?”Jumper回忆起他当时说的话。“如果这不是我们的目标,那我们就着手解决能够真正产生巨大影响的问题。”

凭借在物理学、化学、生物学和计算领域的多重背景,Jumper在头脑风暴会议上分享了独到的见解。很快,他开始领导这个从六人扩展到十五人的团队。“他们在做一件很特别的事情,”Raphael Townshend评价道。2019年,他曾在谷歌DeepMind实习,后来自己成立了AI驱动的生物技术公司Atomic AI。
在学术界,专家们经常各自为政,负责独立的项目,很少有合作。而在DeepMind,统计学、结构生物学、计算化学、软件工程等领域的专家们协同合作,共同解决蛋白质折叠问题。他们还拥有谷歌强大的财政和计算资源支持。“作为博士生,我可能要花几个月的时间才能完成的事情,在这里一天内就能完成,”Townshend说。
他说,伦敦DeepMind办公室氛围活跃,而这种活力很大程度上归功于Jumper。“我觉得他是一个真正的天才,同时也是一个非常谦逊的人,团队成员都非常喜欢他。”计算机科学家Ellen Zhong这样评价道。2021年,她曾在DeepMind实习,现任普林斯顿大学的助理教授。
在Jumper的领导下,团队对AlphaFold进行了重构。他们设计了一种新型的Transformer架构——这是一种深度学习技术,“在过去五年中推动了几乎每一次机器学习领域的突破,”Townshend说道。神经网络通过调整神经元连接的强度来创建更精确的数据表征,主要是关于蛋白质进化和结构的数据。它通过第二个Transformer架构运行这些数据,以预测蛋白质的三维结构。然后,该算法会将预测所得的3D结构与修订后的数据结合起来,返回到Transformer架构中运行数次,以达到提高蛋白质3D结构预测准确性的效果。
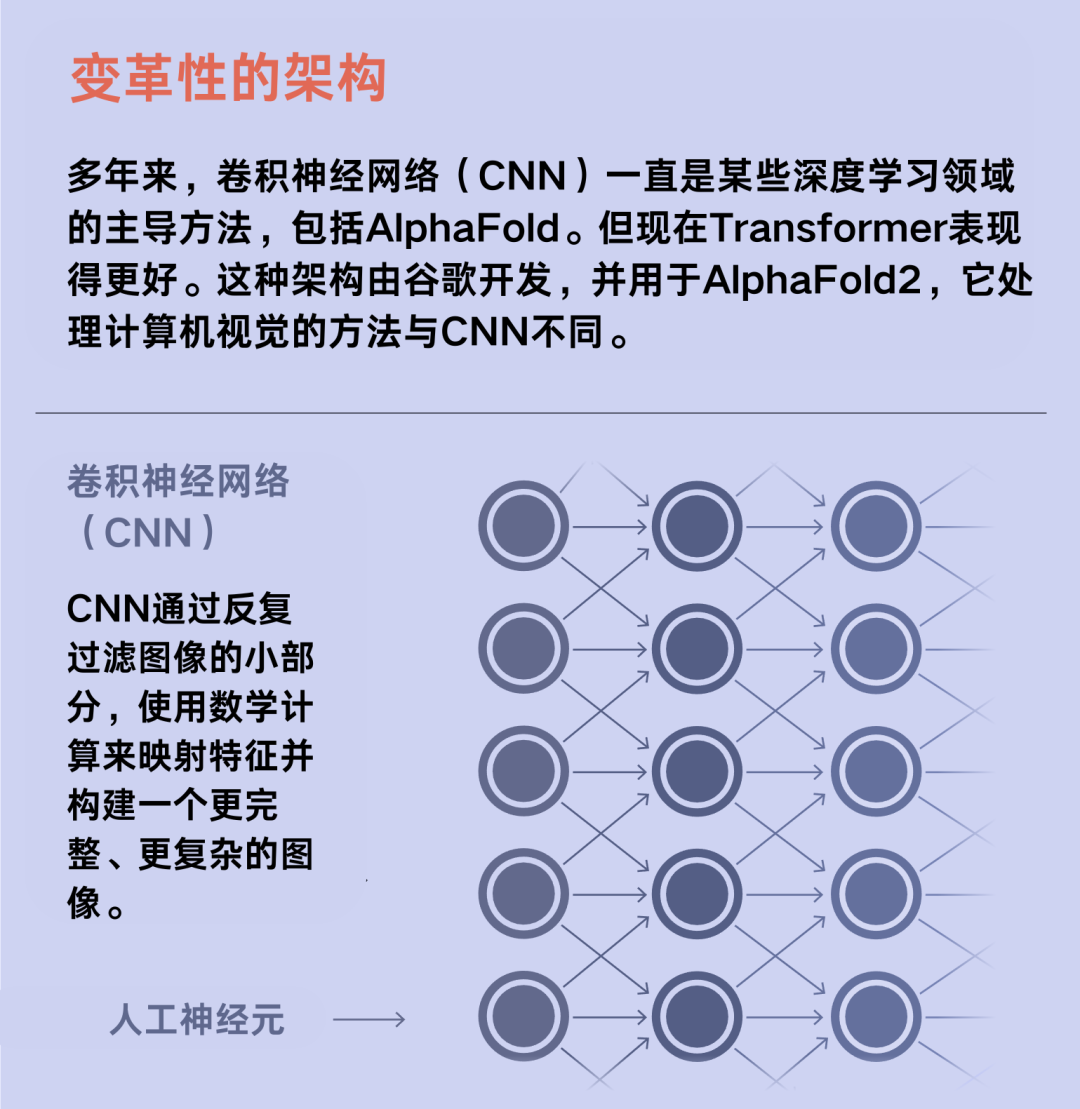
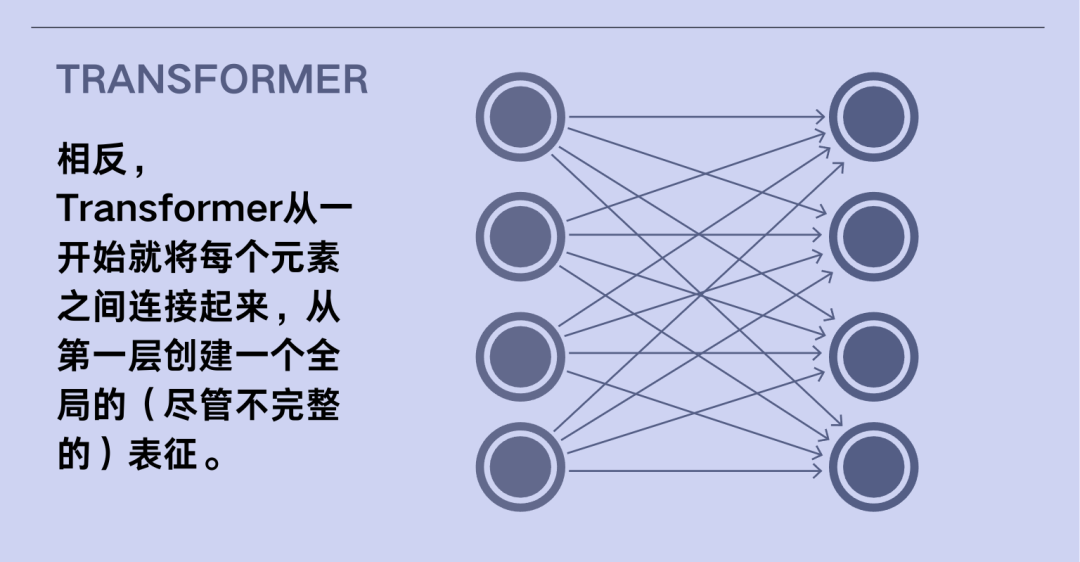
变革性的架构
当他们刚开始研发AlphaFold2时,他们的算法“很糟糕,但没有我们预期的那么糟糕,”Jumper说。“(它)输出了一些看起来有点像蛋白质的螺旋结构。”但随着对算法的进一步优化,他们注意到预测的效率和准确性大幅提升。
“这实际上让人感到害怕,”Jumper说。如果它表现得太好,通常意味着“你做错了一些事情。”但检查过后,他们没有发现问题,优化后的算法就是有效。
团队决定进行一次内部实验,检验他们的系统是否对生物学家有帮助。他们识别出大约50篇发表在《科学》、《自然》和《细胞》等顶级期刊上的论文,这些论文不仅描述了新的蛋白质结构,还基于结构分享了有关蛋白质功能的深刻见解。他们想看看AlphaFold2的效果能否与实验人员费时费力的实验方法相媲美。
他们输入了氨基酸序列,然后开启了AlphaFold2的预测引擎。对于每个输入的序列,AlphaFold2预测出的蛋白质结构都接近论文中实验所得的结果。然而,在团队看来,这还不够准确。这些结构缺少实验人员发现的关键细节。“你感觉好像已经跑完了比赛,却发现其实只完成了一半,”Jumper说道。
接下来的六个月里,团队一点一点的改进系统。距离2020年CASP竞赛的蛋白质候选名单发布还有几周时,他们进行了另一次有用性测试。Jumper对这次测试结果感到满意。于是,谷歌DeepMind在2020年春季向CASP提交了他们的预测效果,然后等待评估结果。
(4)学科大地震
初夏时节,Moult收到了一位CASP评估人员发给他的电子邮件:“看看这个,相当令人印象深刻。”邮件附上了由谷歌DeepMind团队提出的蛋白质结构预测解决方案。Moult确实被震撼到了,但他认为这是单一案例,不可复现。
然后他收到了另一封邮件,然后又一封邮件。“奇怪了,”他回忆自己当时的感受。他收到了三个,四个,许多个接近完美的蛋白质预测结果——而且全部来自DeepMind团队。到了夏末,“我们迅速意识到……发生了非常非常不同寻常的事情,”Moult说道。
CASP的评估人员通过比较预测的蛋白质结构与其经过验证的实验结构来评分。如果模型预测和实验结果一一对应,十分完美,那么得分是100分。Moult一直认为,90以上的分数表明算法有效地解决了蛋白质的结构。AlphaFold的大多数预测结果都达到或超过了90分。
会议前几个月,Moult通过电话将这个消息分享给了Jumper。“我(兴奋地)当场大声咒骂,”Jumper回忆道。“我妻子还关心地问我还好吗。”
2020年12月,在新冠疫情爆发不到一年的时间里,Jumper在CASP在线会议上通过Zoom展示了AlphaFold2。
像其他与会者一样,Jones也在家中观看。“我当时就呆住了……看着眼前发生的一切,”他说。“因为同事不在身边……我们都被封锁在家,无法分享这一切。”
对于任何不是神经网络专家的人来说,这些想法很复杂。即便如此,结论却很明确。DeepMind已经解决了蛋白质折叠问题中的结构预测部分。AlphaFold2能够基于氨基酸序列准确预测蛋白质的结构。
“啊,我的最爱研究课题走到了尽头,”Jones回忆道。“DeepMind终结了比赛,这就是结局。”
多年来,Anastassis Perrakis一直为CASP竞赛贡献未发表的实验结果。当他看到AlphaFold2准确预测出他的团队煞费苦心解析出的蛋白质结构时,他感到心里咯噔一下。
虽然疫情被独自困在家里时,但科学家们一致认为蛋白质科学的世界已经发生了永久性的改变。当这个世界的“居民”眺望眼前的新景色时,他们不由得问出了同一个问题:接下来该怎么办?
3. 余波
3. 余波
(1)惊讶之余,敬畏之心
结构生物学突然失去结构性了。
Silvio Tosatto从CASP创立早期就开始参与相关赛事了,他说,刚开始的时候,很多人陷入了深思。一些结构生物学家担心他们的工作将不复存在,另一些则产生了抵触心理,声称AlphaFold2不够准确。
有些计算生物学家已经为解决蛋白质折叠问题付出了数十年的心血,但当这一刻真的到来时,他们的感受是苦乐参半的。在CASP结束之后的一篇博客文章中,AlQuraishi引用了一位与会者的话,形容自己感觉像是孩子第一次走出家门的父母。

但是,即便对这个耀眼的新工具感到惴惴不安,许多科学家还是非常兴奋。那些不进行结构研究的人过去必须与结构生物学家合作来确定蛋白质结构,现在他们只需按几个按钮就可以自己得出蛋白质的结构。
媒体口中的AlphaFold2成为了“改变一切”的新人工智能突破。但是,科学家们花了数月甚至数年来剖析AlphaFold2的优缺点。Jumper演讲约六个月后,谷歌DeepMind正式发布了这款产品并分享了AlphaFold2的底层代码。“AlphaFold2发布的第二天,我们就尝试将它安装到我们的GPU服务器上了,”Perrakis表示。生物学家们开始了探索。
“我原以为(AlphaFold2)会失败,”Thornton说。“但实际上它却取得了巨大的成功。”

Anastassis Perrakis向CASP贡献了实验蛋白质结构,计算生物学家在其上测试了他们的方法。“我很高兴看到[这些方法]失败,”他开玩笑说。“这不是竞争,但我们喜欢在科学上互相取笑。”
科学家们逐渐意识到,AlphaFold2可能并不会威胁到他们的工作,而是加速研究的催化剂。它不仅没有使结构生物学家失业,反而为他们提供了一种新的工具,使他们的工作更加出色。“如果你认为结构生物学家只是技术专家,他们的工作仅仅是弄清蛋白质的结构,那么他们当然会失业,”Walden说道。但这就像说因为有了人类基因组计划,基因组学家不能再发表描述单个基因序列的论文,所以他们会失业一样。
在许多情况下,结构生物学家的目标其实是探索蛋白质的功能。有了AlphaFold2,他们可以在几分钟内提出假设,而不是等待数月甚至数年来通过实验弄清蛋白质的结构。
Adams认为:“这给结构生物学带来了许多有益的改变,而非坏处,让这个领域更加令人兴奋。”
然而,它并没有立即像一些人预测的那样加速各种新药的研发,因为研究人员很快发现这个工具也有其局限性。Perrakis表示,AlphaFold2的预测并不完美,需要实验验证,但“你可以更快地进入实际的结构研究”。现在,当他的学生开始一个新项目时,他们会首先使用AlphaFold2预测特定蛋白质的结构,然后进行实验验证。
Perrakis认为自己和其他研究人员可能仍会在某些情况下用到X射线晶体学。但是,为了初步构建蛋白质结构,许多人开始将深度学习预测与先进的电子显微镜技术如冷冻电镜(cryo-EM)相结合,这项技术的核心是快速冷冻生物样本并用电子轰击它们。解析了结构之后,他们便可以着手研究蛋白质的功能问题。AlQuraishi表示,AlphaFold2的出现加速了冷冻电镜的应用。
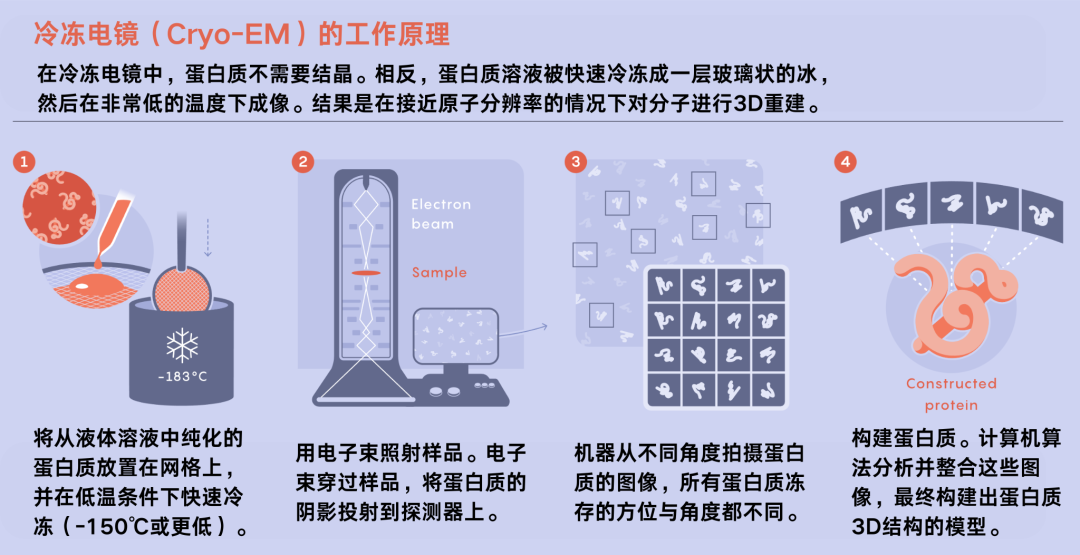
冷冻电镜(Cryo-EM)的工作原理。
这种转变已经开始。2022年6月,《科学》杂志的一期特刊揭示了人类核孔复合体的近原子结构。这个由30种不同蛋白质构成的庞大而复杂的结构几十年来一直是生物学上的难题。科学家们使用AlphaFold2的预测结果来弥补冷冻电镜未能解决的蛋白质结构问题。
Jumper表示,看到这篇论文中的其他科学家使用AlphaFold2取得了生物学上的突破,他在那一刻意识到“(AlphaFold)确实非常、非常重要”。
在过去三年里,像核孔复合体这样的突破标志着蛋白质科学领域的一个又一个里程碑。迄今为止,AlphaFold2预测的蛋白质结构已被用于研究疾病和开发新的药物输送工具。“对我们来说,这非常有帮助,”博德研究所的分子生物学家Feng Zhang表示。他使用AlphaFold2设计了一种将药物输送到人体细胞内的分子注射器。了解蛋白质的结构还可以辅助药物开发,例如,它能帮助研究人员判断哪些分子能够附着在蛋白质结构上,并改变其行为。尽管有些研究表明,AlphaFold2的预测结果在药物研发领域不如实验所得有用,但也有研究证明它可以发挥相同的作用。人们还在探索人工智能工具对药物开发的综合影响。
另一方面,一些生物学家已经不满足于AlphaFold2在识别已知蛋白质结构和功能方面的应用,转向设计自然界中不存在的蛋白质,这种技术对创新药物研发来说十分关键。
(2)新前沿
看到Jumper在2020年CASP会议上的演讲后,Baker立刻回到了他的Rosetta算法的工作上。当时谷歌尚未分享AlphaFold2的底层源代码。尽管如此,“我们开始尝试他们分享的一些想法,”Baker说。谷歌DeepMind团队在《自然》期刊上发表AlphaFold2的当天,Baker和他的团队宣布了RoseTTAFold,这是一种与准确度能与AlphaFold媲美的预测模型。RoseTTAFold同样使用深度学习来预测蛋白质结构,但其底层架构与AlphaFold2大相径庭。
Tosatto表示:“科学理念一旦被提出,其他人就可以逆向工程并在此基础上进行构建,至少那些有足够多资源的人可以做到。”
RoseTTAFold并不是AlphaFold唯一的竞争者。Meta也开发了自己的算法来解决蛋白质结构预测或相关问题。一些公司,已经不局限于蛋白质领域,而是使用深度学习来解析RNA结构,这其中就有Townshend的生物科技初创公司Atomic AI,。然而,在单一结构预测领域,迄今为止还没有人能够达到AlphaFold的精确度,Thornton表示:“我相信有些公司也可以做到,但我认为很难再现AlphaFold当初带来的震撼。”

至少对外而言,Baker和Jumper继续延续着CASP的良性竞争传统。“他们可能觉得我在与他们竞争,但我觉得我们只是从他们的工作中获取灵感,”Baker说道。
Jumper对此表示欢迎,他说:“大家共同推动这门科学的发展真的很重要。如果AlphaFold的智慧结晶后继无人,我觉得是件很悲哀的事情。”
Baker已经扛起了这份传承的重任,并在尝试将之运用到蛋白质科学的新前沿中:蛋白质设计。由于生物学家现在只能研究自然界已经存在的蛋白质,所以Baker设想了一门科学,能够设计出新的蛋白质,专门用来控制阳光、分解塑料或研发新药或疫苗。
荷兰胡布勒支研究所的结构生物学家Danny Sahtoe曾在Baker指导下做博士后研究,他表示:“目前自然界中不同类型的蛋白质结构或形状的数量是相当有限的。理论上,应该有更多可能的形状,而如果你能生成更多形状的蛋白质,那么就意味着你能拥有更多不同功能的蛋白质。”
作为华盛顿大学蛋白质设计研究所的主任,Baker认为蛋白质设计本质上是“逆蛋白质折叠问题”。与其将氨基酸序列输入深度学习算法并让它输出蛋白质结构,蛋白质设计者应该做的是将一个结构输入算法并让它输出一个序列,然后利用这个序列在实验室里生成蛋白质。
AlphaFold和RoseTTAFold本身不能输出这些序列,它们的编程逻辑是相反的。但Baker创建了RoseTTAFold的设计专用迭代版,基于其神经架构,被称为RoseTTAFold diffusion或RF diffusion。
Sahtoe说,蛋白质设计由来已久,但深度学习加速了这一领域的发展,让设计符合现实的蛋白质计算机模型变得“极其快速”。以前,接受过专业培训的蛋白质设计师需要几周或几个月的时间来创建新蛋白质的骨架,但现在他们可以在几天内,甚至一夜之间完成。
Foldit是由Baker实验室开发的在线游戏,玩家可以借此预测蛋白质结构。
Baker还更新了Foldit,将蛋白质设计融入其中。玩家不仅可以构建蛋白质结构,还可以动手设计蛋白质。这个改变颇具成效,Baker的实验室已经发表了几篇关于玩家设计的蛋白质的论文。Baker在华盛顿大学的同事指导的一名研究生就是世界上最顶尖的Foldit玩家之一。
“我们真的理解蛋白质折叠吗?如果我们能够设计出新的序列,并使其折叠成新的结构,那么这表明我们对蛋白质折叠有着相当丰富的了解,”Baker说。“从某种意义上说,这也可以被视为蛋白质折叠问题的解决方案。”
(3)信任练习
AlphaFold2的成功无疑改变了生物学家对人工智能的态度。长期以来,许多实验生物学家不信任计算方法,因为他们知道某些机器学习方法可以使数据看起来比实际更好。但是,谷歌DeepMind团队“明确地证明了你可以把它用在严肃的科研工作中,”AlQuraishi说道。现在如果还有人对此持怀疑态度,其他人便会反驳道:“好吧,那你怎么看AlphaFold呢?”
推动了卷积网络发展的计算生物学家许锦波说:“现在生物学家相信我们的预测结果了,他们以前总是怀疑我们的预测是否可靠。”
这种信任部分源于AlphaFold2平台的一个特性:它不仅生成蛋白质的3D模型,还按照从0到100的信心等级对结构的不同部分进行评分,以此来自我评估预测的准确性。
2022年7月,在谷歌DeepMind发布2180万种蛋白质的结构预测后(几乎涵盖了已知的所有蛋白质)——Adams决定分析AlphaFold2的自我报告。他将预测结果与这些蛋白质的已解结构进行比较,并独立评估其准确性。
“好消息是,当AlphaFold认为自己是正确的时候,它往往确实是对的,”Adams说道。“当它认为自己不对时,现实情况往往也是如此。”但是,在大约10%的情况下,当AlphaFold2对其预测“非常自信”(信心等级得分至少90分)时,其预测结果其实与实验所得有出入,所以它自信过头了。
AI系统似乎有某种自我怀疑的能力,可能会导致人们过度依赖其结论。大多数生物学家仅仅将AlphaFold2视为一种预测工具,但有些人步子迈得太大了。一些以前与结构生物学家合作的细胞生物学家和生物化学家已经用AlphaFold2完全取代了结构生物学家,并将其预测结果奉为圭臬。Perrakis说,有时科学家发表的论文中包含的蛋白质结构在任何结构生物学家看来都显然是错误的。“而他们却说:‘这可是AlphaFold预测的结果。’”

“有些人对这些深度学习模型的能力过于自信,实在太过自信了,”美国国立卫生研究院的研究员Lauren Porter这样认为。“我们应该尽可能多地使用这些深度学习模型,但我们也需要以谨慎和谦逊的态度对待它们。”
Jones听说有科学家想通过计算方法确定蛋白质结构,但在申请资金时却遇到了困难。Jones说:“普遍的看法是DeepMind已经做到了,为什么你还要继续做?”但他认为这项工作仍有必要继续下去,因为AlphaFold2并非完美无缺。
他表示:“(AlphaFold2)与实际科研要求还存在非常大的差距,有些事情它显然无法做到。”
虽然AlphaFold2在预测小而简单的蛋白质结构方面表现出色,但在预测包含多个部分的蛋白质时其准确性较低。它也无法兼顾蛋白质的环境或与其他分子的结合,而这些因素会在自然状态下改变蛋白质的结构。有时,蛋白质需要被某些离子、盐类物质或金属类物质包围才能正确折叠。
Walden说:“目前,AlphaFold还无法确定蛋白质所处的环境。”她的小组已经通过实验确定了几种AlphaFold2无法预测的结构。
AlphaFold2对集中动态蛋白质的预测结果也不是很好,这些蛋白质的功能也非常重要。有些蛋白质的形状会发生改变,因此被称为折叠转换蛋白质(fold-switching protein)。此类蛋白质不是静态的,它们的形状随着与其他分子的相互作用而发生变化。有些即使具有相同的氨基酸序列,也会折叠成截然不同的形状。Porter认为,折叠转换蛋白质“反驳了序列编码单一结构的传统观点,因为这显然与现实不符”。
与DeepMind算法训练用到的数十万种静态单一结构蛋白质相比,折叠转换蛋白质只有大约100个,当然肯定还有更多尚未被发现。Porter说,其实这可能也不是很出人意料,因为“一般来说,这些算法就是为了预测单一折叠而设计的”。
还有一些内在无序蛋白质或蛋白质区域缺乏稳定结构,就像是汽车经销商门口摆来摆去的充气玩偶,总是在不停的扭动、重组。“它们在很多方面被忽视了,因为它们有点烦人,”哥本哈根大学计算蛋白质生物物理学教授Kresten Lindorff-Larsen说道。大约44%的人类蛋白质存在至少一个由30个氨基酸组成的无序区域。Lindorff-Larsen表示:“这是一个相对较大的比例。”
AlphaFold2可以预测某些区域何时会内在无序的情况,但它无法告诉你这种无序的具体形态。
对于Jumper来说,最令他失望的是AlphaFold2无法区分两个仅相差一个氨基酸的蛋白质,也被称为点突变(point mutation)。他说,点突变有时会对蛋白质的结构和功能产生相当显著的影响,但“AlphaFold却对它们几乎视而不见,”基于两段不同的序列生成相同结构预测。
2023年9月,DeepMind发布了AlphaMissense,这是一种预测此类突变影响的深度学习算法。它不能显示结构的变化,但它可以根据已知病原蛋白中的类似突变,告知用户该突变是否可能使蛋白质致病或诱发功能障碍。
然而,即使AlphaFold2能够完美预测所有蛋白质,它也离模拟现实中的生物原理相距甚远。因为在细胞中,蛋白质从不单独行动。
(4)细胞复杂性
细胞内部复杂且混乱。细胞的外膜包裹着一个拥挤的生化环境,其中充满了各种分子成分——蛋白质、信号分子、信使RNA、细胞器等。蛋白质相互结合并与其他分子结合,这改变了它们的形式和功能。
AlphaFold2预测单个蛋白质结构的能力并没有帮助生物学家更好理解复杂天然环境中的蛋白质。但是,这正是该领域目前正在前进的方向。蛋白质科学领域的人工智能巨头——谷歌DeepMind和David Baker的蛋白质设计研究所,都在优化它们的深度学习算法,以预测蛋白质在与其他分子相互作用时所呈现的结构。
在2024年春天,他们都发表了有关在该领域取得相似进展的论文。通过更新算法,他们分别发布了AlphaFold3和RoseTTAFold All-Atom,它们能够预测蛋白质与其他蛋白质、DNA、RNA和其他小分子结合时的结构。
生物学家也开始对这些新模型进行测试。目前为止,AlphaFold3比RoseTTAFold All-Atom准确许多,AlQuraishi表示,但这不是像“AlphaFold2”那样的技术飞跃。对于某些大分子,如RNA结构,其准确性仍低于其他基于物理学的系统和实验。
尽管如此,这些新算法朝着正确的方向迈进了一步。蛋白质与其他分子的相互作用对其在细胞中的功能至关重要。为了开发可以与蛋白质结合并按需改变其活性的药物,研究人员需要了解这些复合物的形态。然而,Adams说,任何一种算法在短时间内都不太可能催生新的药物。“这两种方法的准确性仍然有限,(但)它们都在力所能及的范围内实现了巨大进步。”
DeepMind的新产品还发生了一个重要的变化。此前AlphaFold2的底层代码是开源的,其他研究人员可以研究该算法,在此基础上进行调整,以便更好地服务自己的项目。然而,谷歌迄今为止都未公开AlphaFold3的源代码,而是将其作为商业机密加以保护。AlQuraishi说:“至少目前,没有人可以像使用(AlphaFold2)那样运行和使用它。

即使在AlphaFold3发布之前,研究人员就已经在测试AlphaFold2,看看它是否能提供有关不同构象下蛋白质的有用信息。布朗大学化学和物理学副教授Brenda Rubenstein对激酶(kinases)这种能激活其他蛋白质的蛋白质很感兴趣。具体来说,她想了解一种诱发癌症的激酶的机制,以此开发更精准对症的药物。她的实验室使用物理学的方法对激酶的结构进行了建模,即基于牛顿定律绘制原子的3D坐标。这个过程花了两年半的时间。
Rubenstein说:“大约一年前,我们开始思考能否加速这个过程?”他们尝试以一种创新的方式使用AlphaFold2。通过向算法输入有关相关蛋白质的数据,她发现其预测不同构象激酶的准确度超过了80%。
AlQuraishi评价道,Rubenstein的实验室为数不多能够发现“如果你以正确的方式‘刺激’AlphaFold,它会输出多个构象”的实验室之一。“这令人鼓舞。”
AlQuraishi希望到2040年,深度学习能够模拟一个完整的细胞及其内部的所有结构和动态。然而,要实现这一目标,需要在实验和计算两方面取得质的飞跃。
(5)非专业视角
对于许多生物学家来说,AlphaFold2是他们一直在等待的突破性技术。CASP的目标是创建能够基于氨基酸序列预测蛋白质结构的计算工具。然而,许多人不禁要问:为什么一个初出茅庐的新手能够破解蛋白质的奥秘,而那么多专家却苦苦挣扎了几十年?
不可否认的是,谷歌DeepMind的计算机和蛋白质科学团队为解决这个问题提供了新的视角。同时,蛋白质科学已有无数先驱为其打下了坚实的基础,做好了迎接深度学习革命的准备。AlQuraishi说:“这些突破不是凭空出现的。”
到2020年CASP启动时,许多研究人员预计结构预测的突破将通过人工智能实现。“一切都朝着这个方向发展,”Townshend 说。但他们没有想到会来自一家市值数十亿美元的科技公司,也没有想到会这么快。有些人认为AlphaFold2并不是全新的科学成就,而是精妙的工程技术;有些人对David Baker的算法没有获奖感到惊讶;还有些人则认为谷歌DeepMind坐拥其他公司和团队无法匹敌的资源,因此不足为奇。
每年大约有100个实验室参加CASP,尽管他们已经开始采用AI技术,但他们“可能没有DeepMind的专业人才储备,也没有同等的算力,”Thornton这样认为。DeepMind“可以使用的算力几乎是无限的”。
她还推测,谷歌虽然在蛋白质科学方面缺乏专业知识,但这反而可能释放了他们的创造力。“他们专注于建立一个强大的神经网络,”Thornton说。蛋白质生物学家包袱更重。当他们开发AI工具时,他们希望捕捉蛋白质折叠过程中涉及的原子级分子物理和化学原理。DeepMind的想法不一样:我们只需序列数据转化为3D结构,至于如何实现,那无所谓。
Walden说:“他们没有像此前很多预测模型那样,试图解决蛋白质折叠的问题本身,而是单纯用‘蛮力’”去绘制原子在空间中的最终位置。有趣的是,他们反而因此解决了这个问题。”

对一些生物学家来说,这种方法并没有完全解决蛋白质折叠问题。结构生物学创立的初心是希望了解氨基酸链是如何折叠成蛋白质的。大多数生物学家认为AlphaFold2确实解决了结构预测问题,但却没有解决蛋白质折叠问题。Ellen Zhong表示:“现在,我们只是拥有了一个黑匣子,它可以某种方式告诉你最终的折叠状态,但实际上你却不知道是如何实现的。”
“这不是科学家解决问题的方式,”布朗大学计算机科学家Littman表示。
但约翰霍普金斯大学生物物理学荣誉教授George Rose表示,这听起来可能像是在‘找茬’,但显然不是。”AlphaFold2可以根据对数十万蛋白质结构的分析,识别出给定氨基酸序列潜在折叠模式。但它无法告诉科学家蛋白质折叠过程的任何信息。
“对于许多人来说,你不需要知道。他们不在乎,”Rose说。“但科学,至少从过去500年以来……一直致力于理解事物发生的过程。”为了理解以蛋白质为基础的生命的动态变化、机制、功能和本质,Rose认为,你需要具备对过程的全面了解——而深度学习算法无法做到这一点。
对Moult来说,重要的并不是机器在做的事情他无法理解。“我们都习惯了机器做我们无法做到的事情。比如,我跑得不如我的车快,”他说。对于想要研究蛋白质的分子生物学家来说,只需要大致知道它的结构即可,至于蛋白质是如何折叠形成这样的结构并不重要。
但“在我们真正知道它是如何运作之前,我们永远不会有一个100%可靠的预测器,”Porter说。“我们必须理解基本的物理学,才能做出最有根据的预测。”
AlQuraishi 表示:“我们的目标在不断变化。我确实认为核心问题已经解决,现在更加关心接下来会发生什么。”
即使生物学家之间还存在不少争辩,其他人已将目光投向了一个毫无疑问已经改变的领域,并开始回顾起近期的发展。
有时Perrakis会怀念以前的工作方式。2022年,他的团队通过X射线晶体学解析了一种参与修饰微管的酶的结构(微管microtubules)是形成细胞结构的巨大棒状分子。他说:“我意识到我再也不会(这样做)了,也无法再次感受到在数月的工作之后首次确定结构带来的特殊满足感。”
AlphaFold2并没有淘汰这些实验。相反,它揭示了它们的必要性。它将两个历史上截然不同的学科联系在一起,触发了一场酣畅淋漓的对话。
(6)新世界
七十年前,人们认为蛋白质是一种胶状物质,Porter说。“而现在我们能看到的”:一个由各种结构构成的巨大的蛋白质世界,无论它们是自然界中存在的还是设计出来的。
蛋白质生物学领域“在AlphaFold出现之后更令人兴奋了”,Perrakis说。这种兴奋来自于基于蛋白质结构促进药物发现的可能,来自各种假说的层出不穷,来自理解细胞内复杂相互作用的希望。
“这感觉就像基因组学革命一样,”AlQuraishi说。面对海量数据,无论是在湿实验室还是在计算机前的生物学家都在学习如何处理这些数据。
但就像在全球范围内激发的其他人工智能突破一样,这可能也有一个天花板。
AlphaFold2的成功建立在大量可用的高质量训练数据上,也就是勤勤恳恳的实验人员通过一丝不苟的工作解析的数十万种蛋白质结构。虽然AlphaFold3和相关算法在确定分子化合物结构方面取得了一些成功,但其准确性仍不及对单蛋白质的预测。有部分原因就是因为可用的训练数据少了很多。
Thornton 表示,蛋白质折叠问题“几乎是一个AI解决方案的完美例证”,因为相关算法的训练数据来自以统一形式收集的数十万种蛋白质结构。然而,蛋白质数据库可能是生物学领域有组织的数据共享的特殊案例。没有高质量的数据来训练算法,AI模型将无法做出准确的预测。
“我们很幸运,”Jumper说。“我们遇到问题的时候恰逢解决问题的时机成熟。”
没有人知道深度学习在解决蛋白质折叠问题上的成功是否会延续到其他科学领域,甚至是生物学的其他领域。但有些人,比如AlQuraishi,持乐观态度。“蛋白质折叠只是冰山一角,”他说。例如,化学家需要进行的计算成本高昂。通过深度学习,这些计算已经比以前快了一百多万倍,AlQuraishi说。
人工智能显然可以推进特定类型的科学问题的解决。但它可能只能增进科学家对结果的了解,而AlQuraishi表示:“从历史的角度出发,科学的本质在于理解原理,”也就是生命和宇宙演化的基本过程。如果科学通过给出解决方案而不是揭示过程的深度学习工具向前发展,它还能称得上是科学吗?
“如果你能治愈癌症,你真的在乎它是如何被治愈的吗?”AlQuraishi说。“这是一个我们将在接下来数年里反复争论的问题。”
如果许多研究人员决定放弃理解自然的过程,那么人工智能不仅会改变科学——它还会改变科学家。
与此同时,CASP的组织者正在处理另一个问题:如何继续他们的竞赛和会议。AlphaFold2是CASP催生的产物,它解决了会议旨在解决的主要问题。“对我们来说,这在某种程度上是一个巨大的冲击:CASP现在要何去何从?”Moult说。
2022 年,CASP会议在土耳其安塔利亚举行。谷歌DeepMind团队没有参赛,但存在感依然强烈。“几乎是AlphaFold使用者之间的比赛,”Jones说。从这个意义上说,最大赢家还是谷歌。
现在一些研究人员对参加会议的兴趣减少了。“看到那个结果后,我改变了我的研究方向,”许锦波说。其他人则继续完善他们的算法。Jones仍然继续结构预测研究,但这对他来说现在更像是一种爱好。还有一些人,比如AlQuraishi和Baker,继续开发新的结构预测和设计算法,即便是与一家市值数十亿美元的公司竞争也丝毫不怵。
Moult和会议组织者正在尝试与时俱进。下一轮CASP于五月开始接受报名。他希望深度学习能征服结构生物学的更多领域,比如RNA或生物分子复合物。“这种方法解决了一个问题,”Moult说。“但在结构生物学中还有许多其他相关问题。”
下一次会议将于2024年12月在加勒比海举行。蓝天碧海,风和日丽,相信大家的交流也会在友好的氛围中进行。至少现在,没有人会再公开跺脚以示反对了。谁也猜不到今年的比赛会是什么样子。但基于往届CASP的经验来看,Moult知道有一件事是大家可以期待的:“惊喜。”
原文链接: https://www.quantamagazine.org/how-ai-revolutionized-protein-science-but-didnt-end-it-20240626/
生命复杂性读书会招募中
在生物学中心法则的起点,基因作为生命复杂系统的遗传信息载体,在生命周期内稳定存在;而位于中心法则末端的蛋白质,其组织构成和时空变化的复杂性呈指数式增长。随着分子生物学数十年来的突飞猛进,尤其是生命组学(基因组学、转录组学、蛋白质组学和代谢组学等的集合)等领域的日新月异,当代生命科学临近爆发的边缘。如此海量的数据如何帮助我们揭示宇宙中最复杂的物质系统——“人体”的构成原理和设计原理?阐释人类发育、衰老和重大疾病的发生机制?
集智俱乐部联合西湖大学理学院及交叉科学中心讲席教授汤雷翰,国家蛋白质科学中心(北京)副研究员常乘、李杨,香港浸会大学助理教授唐乾元,北京大学前沿交叉学科研究院研究员林一瀚,中国科学院分子细胞科学卓越创新中心博士后唐诗婕,共同发起「生命复杂系统的构成原理」读书会,从微观细胞尺度、介观组织器官尺度到宏观人体尺度,梳理生命科学领域中的重要问题及重要数据,由生物学家提问,希望促进统计物理、机器学习方法研究者和生命科学研究者之间的深度交流,建立跨学科合作关系,激发新的研究思路和合作项目。读书会从2024年8月6日开始,每周二晚19:00-21:00进行,持续时间预计10-12周。欢迎对这个生命科学、物理学、计算机科学、复杂系统科学深度交叉的前沿领域感兴趣的朋友加入!
详情请见:
生命复杂性读书会:从微观到宏观,多尺度视角探索生命复杂系统的构成原理
大模型与生物医学:
AI + Science第二季读书会
详情请见:
推荐阅读
1. 如何发现 AI+Science 中的下一个 AlphaFold 和 ChatGPT?
2. Nature速递:AlphaFold 3 预测所有生命分子的结构和相互作用
3. 2亿个AlphaFold预测结构中隐藏的蛋白质进化趋势 | 集智科学家最新成果
4. 张江:第三代人工智能技术基础——从可微分编程到因果推理 | 集智学园全新课程
5. 龙年大运起,学习正当时!解锁集智全站内容,开启新年学习计划
点击“阅读原文”,报名读书会
微信扫一扫,分享到朋友圈











