在生命体系中,蛋白质是执行各种生物功能的主要生物大分子。蛋白质分子和由蛋白质分子组成的复合系统如同宏观世界中不同的机器一样,它们都会发生运动、改变其构象或结构,然后行使复杂的生物功能。理解和预测蛋白质的动力学信息是了解其生物功能以及设计新药物的基础,是当前分子生物学具有挑战性的难题。尽管当前以AlphaFold为代表的许多AI算法在蛋白质天然态稳定结构预测取得了巨大的进展,但还难以准确捕捉蛋白质的结构变化信息。
近期,南京大学王炜教授、李文飞教授团队(论文第一作者管星悦,南京大学与国科温州研究院联合培养博士生)与美国国家科学院院士、莱斯大学Peter G. Wolynes教授,香港浸会大学物理系唐乾元助理教授,国科温州研究院任卫同副研究员,昌平实验室领衔科学家陈明辰博士等人合作,提出结合物理学原理解决基于AlphaFold预测蛋白质动力学的一个新方案,发展了利用蛋白质能量面信息来驱动人工智能预测蛋白质动力学的新方法,准确预测了典型蛋白质的结构变化及其转变路径。提出的研究方案,为基于AI研究蛋白质动力学的挑战性问题提供了解决途径,亦为物理学与人工智能紧密结合研究分子生物学提供了一个成功范例。相关研究成果近日发表在美国科学院院刊 PNAS 上。
研究领域:蛋白质动力学,阻挫最小原理,蛋白质结构预测,生物物理,生物信息学,人工智能
管星悦、唐乾元、任卫同、李文飞 | 作者
王炜 | 审校

Predicting protein conformational motions using energetic frustration analysis and AlphaFold2
https://www.pnas.org/doi/10.1073/pnas.2410662121
1. 基于AlphaFold 2 的蛋白质结构预测
近年来,随着人工智能(AI)技术的发展,基于序列的蛋白质结构预测问题取得了许多重要的突破。其中,由 DeepMind 开发的AlphaFold 2 (以下简称为AF2),被誉为AI领域的一项革命性进展,在部分蛋白质的结构预测任务中,其对天然态三维静态结构预测的准确性逼近实验结果[1]。
为什么AF2能够如此成功地预测蛋白质的结构呢?这是因为AF2利用基于注意力机制(Attention Mechanism)的Transformer架构,成功从蛋白质的共进化信息中揭示了蛋白质内部氨基酸之间的空间关联。如果蛋白质序列中两个位点的残基倾向于协同突变,则这两个位点通常在空间上也非常接近且存在相互作用:这也是被充分验证的生物物理知识。AF2利用大量的与待预测序列相似的序列(即所谓“同源序列”),对这些序列进行多序列比对(Multiple Sequence Alignment, 以下简称为MSA),基于MSA空间中所蕴藏的共进化信息,得出蛋白质内部残基间的关键约束,在此基础上实现了成功的结构预测。简而言之,AF2的成功是生物物理知识与强大的神经网络框架的合作成果。
然而,蛋白质为了执行功能,往往需要在多个不同的结构之间产生动态变化,这种过程被称为“构象变化”或者“构象运动”。尽管AF2在蛋白质的天然态三维静态结构预测方面取得了许多重要的成果,但其难以给出对蛋白质结构动态变化的预测。为解决蛋白质构象变化的预测问题,有两种不同的思路:
-
一种是“打开黑箱”,直接对AF2的基本框架进行改造,这一方法涉及对AF2模型结构、参数调整、训练方法等方面的改造,如果这种改造成功,毫无疑问将显著增强AF2在复杂生物学问题上的应用潜力;
-
而另一种则是“维持黑箱”,基于现有的AF2框架,对AF2的输入信息进行微调(给出不同的“提示”),从而使AF2预测出蛋白质的构象变化。
毫无疑问,后一种思路的训练成本低、易于操作,同时该方法能更大程度上直接建立序列改变与结构改变之间的定量联系,为进一步的蛋白质理性设计和药物设计提供了更加灵活且可行的解决方案。本研究正是基于后一种研究思路而展开的,相关的开发基于ColabFold [2]。
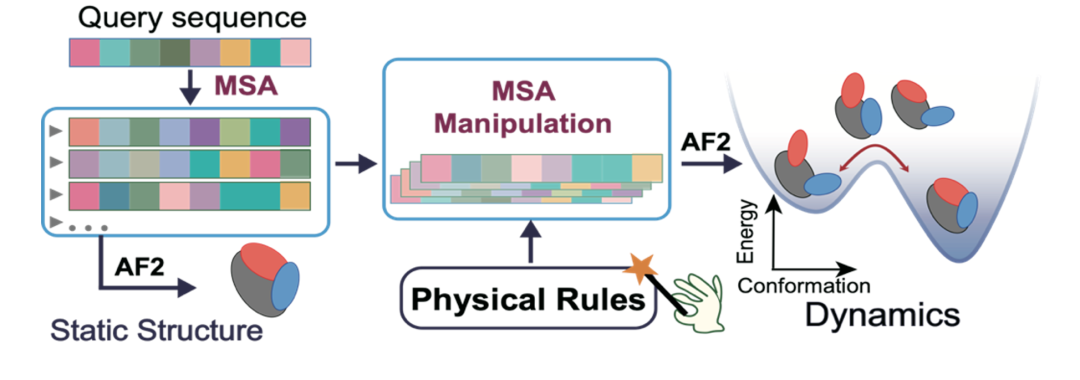
为了获取蛋白质构象运动的信息,我们需要预测同一蛋白质在不同构象状态下的结构。在这种情况下,保持输入的序列不变,我们仍然可以通过筛选前文提到的MSA空间中的序列来影响预测结果。此前,有研究表明,通过对多序列比对进行序列相似性聚类(AF Cluster),可以使AF2对已知的变构蛋白的不同构象状态进行采样 [3]。这一发现为我们利用AF2预测蛋白质的构象变化提供了新的思路:即通过筛选MSA空间中所包含的序列集合,改变相关的共进化提示,可以诱导模型预测出蛋白质的不同构象状态(如图1所示)。与AF Cluster等其它基于序列筛选的方法的不同点在于,我们希望在序列筛选的过程中引入更多物理知识的指导,从而准确识别调控蛋白质构象变化的关键残基,并实现预测结果可解释。
图 1:研究方法示意图:根据物理知识筛选MSA中的序列,引导AF2预测蛋白质的不同构象状态。
美国国家科学院院士、莱斯大学的Peter G. Wolynes教授(本文的合作者之一)及其同事在20世纪80年代提出的蛋白质能量面理论,为预测蛋白质结构与动力学提供了基本的物理学原理:蛋白质折叠可以被看作是在漏斗状的能量面内下降至能量最低点的过程,而这个最低点对应蛋白质最稳定的天然结构。Wolynes等人还提出了蛋白质折叠中的阻挫最小原理(principle of minimal frustration):即蛋白质的序列在进化中不断被优化,使得在能量面上较少出现局部极小值或能量障碍,进而有助于蛋白质更容易地找到其全局能量最低的天然构象 [4]。这一原理解释了为什么大多数天然蛋白质可以在细胞环境中迅速且高效地折叠成其特定的三维结构,同时减少错误折叠的概率。
然而,天然态蛋白质为了执行功能,仍需灵活地发生构象运动,因此其局部能量并不是最优化的,而是保留了一定的“冲突”与不稳定性,这种现象被称为“局部阻挫”(local frustration)。正是由于这些局部阻挫的存在,天然态蛋白质表现出“总体稳定而局部灵活”的特征,这是一种在天然态结构稳定性与功能所需的运动灵活性之间的平衡,并被自然界的酶分子等别构蛋白所采用,作为实现高效率功能运动的关健物理策略[4-7]。要精准刻画指导蛋白质构象引动的能量面特征,局部阻挫信息无疑起着至关重要的作用。
利用现有的阻挫分析工具 Frustratometer [8],我们可以识别蛋白质哪部分区域有较高的局部阻挫并因此容易引发结构变化 [5]。根据识别出的高阻挫位点或区域,我们就得以筛选和操纵MSA空间中所包含的序列及其所蕴藏的共进化信息——这些共进化信息正是AF2用以预测天然结构的重要提示依据。通过提供不同的共进化信息,即使是基于同样的输入序列,我们也可以利用AF2得到不同的预测结构,从而能够准确预测蛋白质的构象变化路径。
3. 应用案例:
预测腺苷酸激酶的变构运动中的亚稳态
下面将以腺苷酸激酶(AdK)为例,简要说明上述研究思路和方法,更多具体结果及其它案例请参考论文原文。AdK蛋白在细胞能量代谢中起着核心作用,催化ATP、ADP和AMP之间的转换,维持细胞的能量代谢平衡。AdK蛋白有Core、Lid、NMP三个结构域,为了完成催化,AdK必须在Closed态和Open态之间转换,而Closed态对应AF2的默认预测结构,即被AF2判定为最稳定的基态结构。在AF2预测出了Closed态结构的基础上,我们的目标则是找到AdK的Open态及其变构路径。
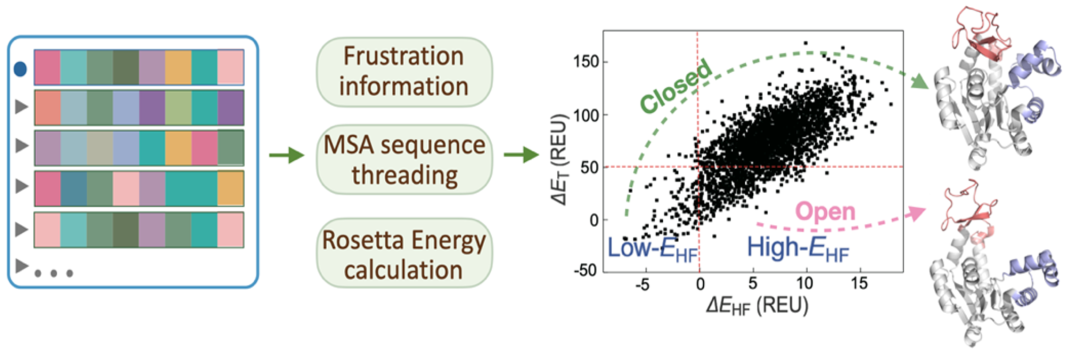
基于AF2的Closed态结构,可以使用Frustratometer [8] 分析AdK蛋白的局部阻挫。研究表明变构过程中发生大尺度构象变化的位点以及断开的相互作用连接处往往具有更高的阻挫。在识别了高阻挫位点之后,我们将MSA空间中的各条序列替换到AF2给出的AdK蛋白Closed态结构上,用Rosetta计算能量变化,具体来说,我们主要关注(1) 高阻挫位点的能量变化以及(2)总能量变化。在总能量相对稳定的前提下,我们发现,使高阻挫位点保持稳定的MSA序列能够引导AF2给出能量稳定的Closed态(与AF2默认预测结构相同),而使高阻挫位点不稳定的MSA序列则会引导AF2的预测结果偏离基态而前往亚稳态,给出变构之后的Open态结构(如图2所示)。
图 2:结合阻挫信息对MSA空间中的序列进行筛选,能够使AF2预测蛋白质的亚稳态结构。
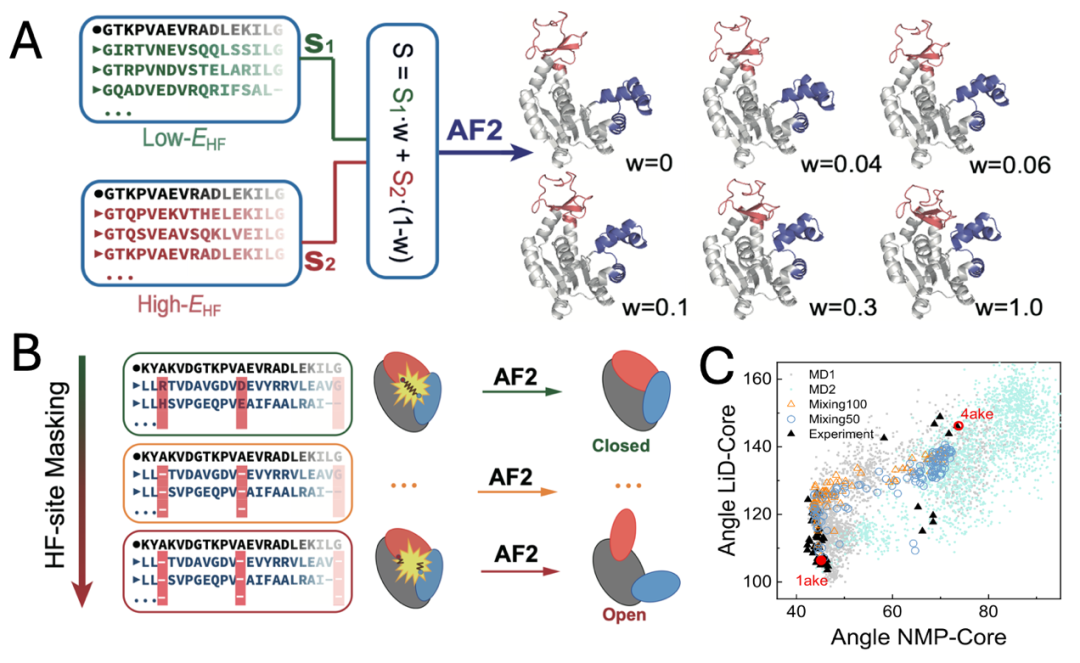
通过更细致地调节高阻挫位点上的不稳定性,还可以得到AdK蛋白的变构路径细节。例如,我们可以采取序列混合法:从高阻挫位点稳定序列集(图2中Low-EHF区域)与高阻挫位点不稳定序列集(图2中High-EHF区域)分别按照一定的比例随机抽取序列并混合生成新的MSA序列集(如图3A所示),或者在上述两个区域的序列空间之间构建一条能量连续变化的“路径”,将重采样的序列集进行AF2结构预测。随着高阻挫位点不稳定序列比例的增加,所预测出的结构逐渐由Closed态向Open态连续转变,从而给出变构路径的预测。
此外,另一种在高阻挫位点处引入不稳定性的方法则是位点遮盖法:通过将MSA空间中序列的某一位点替换为缺省值,屏蔽了该位点的信息,避免AF2识别到相关位点附近的共进化信息(如图3B所示)。该方法同样可以使得预测结果偏离基态而给出亚稳态结构。值得注意的是,在低阻挫位点引入同样的缺省值并不能带来变构。
用上文提到的两种方法都可以成功得到AdK的变构路径。如图3C所示,用Lid、NMP两个结构域与Core结构域分别所成的夹角来衡量变构路径,所预测的变构路径与实验及分子模拟结果一致。该方法甚至可以根据需求而给出蛋白质不同于变构路径的独特结构,相关的探索对蛋白理性设计问题具有重要参考意义。
以上结果表明,基于蛋白质能量面理论,识别蛋白质的高阻挫位点,并采用各种方法在MSA空间理性筛选共进化信息,不仅能够让AF2突破其限制,预测出蛋白的亚稳态结构,还能得到变构路径等关键信息。
图 3:(A)序列混合法与(B)位点遮盖法示意图,(C)本方法所预测的AdK蛋白Closed-Open的变构路径。
本研究表明,通过将支配蛋白质运动的物理规则与预测蛋白质结构的人工智能相结合,我们能够准确预测蛋白质的结构变化和运动路径。我们的方法在蛋白质序列、能量阻挫、共进化信息与蛋白质结构和动力学之间建立了有机联系,为蛋白质设计等重要问题提供了新的启发。
尽管人工智能在生物物理领域的突破展示了数据驱动方法的强大能力,但我们的研究表明,在后AF2时代,基于物理原理的研究方法仍然具有不可替代的重要性。这一工作是 AI for Biosciences 领域的一个成功应用,不仅解决了在人工智能预测蛋白质动力学领域中的一个挑战性问题,更是为物理与人工智能的结合开辟了新的途径。
本研究展示了将生物物理知识与人工智能结合的创新方法在未来研究和实际应用中的广泛潜力。特别是在药物设计、酶工程和疾病机制解析等领域,这种跨学科的结合能够带来具体的应用进展。例如,它可以帮助设计更有效的药物分子、优化酶的功能,揭示疾病发生的分子机制。未来,物理与人工智能的紧密协作,有望推动这些领域的发展,带来更精准的生物分子设计,帮助我们更深入地理解复杂的生命过程。
本项目研究得到了国家自然科学基金(NSFC:11974173, 11934008, 12305052)、中国科学院大学温州研究院(WIUCASQD2021010)、香港研究资助局(RGC:22302723)等的支持。
1. Jumper, J. et al. “Highly accurate protein structure prediction with AlphaFold.” Nature, 596, pages 583–589 (2021). DOI: 10.1038/s41586-021-03819-2
2. Mirdita, M. et al. “ColabFold: Making protein folding accessible to all.” Nature Methods, 19, pages 679–682 (2022). DOI: 10.1038/s41592-022-01488-1
3. Wayment-Steele H K, et al. “Predicting multiple conformations via sequence clustering and AF2”, Nature, 625, pages 832–839 (2024). DOI: 10.1038/s41586-023-06832-9
4. Ferreiro, D. U., Komives, E. A., & Wolynes, P. G. (2014). Frustration in biomolecules. Quarterly Reviews of Biophysics, 47(4), 285-363. DOI: 10.1017/S0033583514000092
5. Li, W., Wolynes, P. G., & Takada, S. (2011). Frustration, specific sequence dependence, and nonlinearity in large-amplitude fluctuations of allosteric proteins. Proceedings of the National Academy of Sciences, 108(9), 3504-3509. DOI: 10.1073/pnas.1018983108
6. Li, W., Wang, J., Zhang, J., Takada, S., & Wang, W. (2019). Overcoming the bottleneck of the enzymatic cycle by steric frustration. Physical Review Letters, 122(23), 238102. DOI: 10.1103/PhysRevLett.122.238102
7. Zhang, Y., Chen, M., Lu, J., Li, W., Wolynes, P. G., & Wang, W. (2022). Frustration and the kinetic repartitioning mechanism of substrate inhibition in enzyme catalysis. The Journal of Physical Chemistry B, 126(36), 6792-6801. DOI: 10.1021/acs.jpcb.2c03832
8. Parra R. G., et al. “Protein Frustratometer 2: a tool to localize energetic frustration in protein molecules, now with electrostatics”, Nucleic Acids Res. 44(Web Server issue): W356–W360 (2016). DOI: 10.1093/nar/gkw304
1. 陈明辰博士为昌平实验室领衔科学家,课题组正在积极招聘AI for Biomolecular Science的各层级的有志之士,欢迎联系邮箱:mingchenchen@cpl.ac.cn。
2. 香港浸会大学物理系唐乾元课题组(傅渥成)正在招收统计物理、机器学习、生物信息学、计算神经科学及相关领域的博士生、研究助理、博士后,欢迎联系邮箱:tangqy@hkbu.edu.hk 。
在生物学中心法则的起点,基因作为生命复杂系统的遗传信息载体,在生命周期内稳定存在;而位于中心法则末端的蛋白质,其组织构成和时空变化的复杂性呈指数式增长。随着分子生物学数十年来的突飞猛进,尤其是生命组学(基因组学、转录组学、蛋白质组学和代谢组学等的集合)等领域的日新月异,当代生命科学临近爆发的边缘。如此海量的数据如何帮助我们揭示宇宙中最复杂的物质系统——“人体”的构成原理和设计原理?阐释人类发育、衰老和重大疾病的发生机制?
集智俱乐部联合西湖大学理学院及交叉科学中心讲席教授汤雷翰,国家蛋白质科学中心(北京)副研究员常乘、李杨,香港浸会大学助理教授唐乾元,北京大学前沿交叉学科研究院研究员林一瀚,中国科学院分子细胞科学卓越创新中心博士后唐诗婕,共同发起「生命复杂性:生命复杂系统的构成原理」读书会,从微观细胞尺度、介观组织器官尺度到宏观人体尺度,梳理生命科学领域中的重要问题及重要数据,由生物学家提问,希望促进统计物理、机器学习方法研究者和生命科学研究者之间的深度交流,建立跨学科合作关系,激发新的研究思路和合作项目。读书会从2024年8月6日开始,每周二晚19:00-21:00进行,持续时间预计10-12周。欢迎对这个生命科学、物理学、计算机科学、复杂系统科学深度交叉的前沿领域感兴趣的朋友加入!
6. 加入集智,一起复杂!