Nature Methods特刊评论:用人工智能之“钥”,开空间组学之“锁”


文章标题:Unlocking the power of spatial omics with AI
文章来源:Nature Methods
文章链接:https://www.nature.com/articles/s41592-024-02363-x
空间组学技术通过提供详细的、空间定位的分子特征,极大地改变了生物医学研究范式,使我们能够以前所未有的分辨率阐明组织结构和功能。人工智能(AI)有可能充分释放空间组学的潜力,促进复杂数据集的整合并发现新的生物医学结论。
——卷首语
空间组学技术开启了生物医学研究的新纪元,利用空间分子特征,我们能够对组织结构进行前所未有的精细洞察。众多空间组学技术使我们能够利用各种组学和成像方式分析组织切片,从而更全面地探索组织结构。尽管取得了显著进展,但不同数据类型的整合仍面临巨大挑战,同时也需要克服技术本身固有的局限性。在此评论中,我们讨论了生成和分析空间组学数据的关键瓶颈,并强调了AI如何提出有望将这些挑战转化为机遇的方法,以充分利用空间组学的力量推动进一步的突破(图1)。
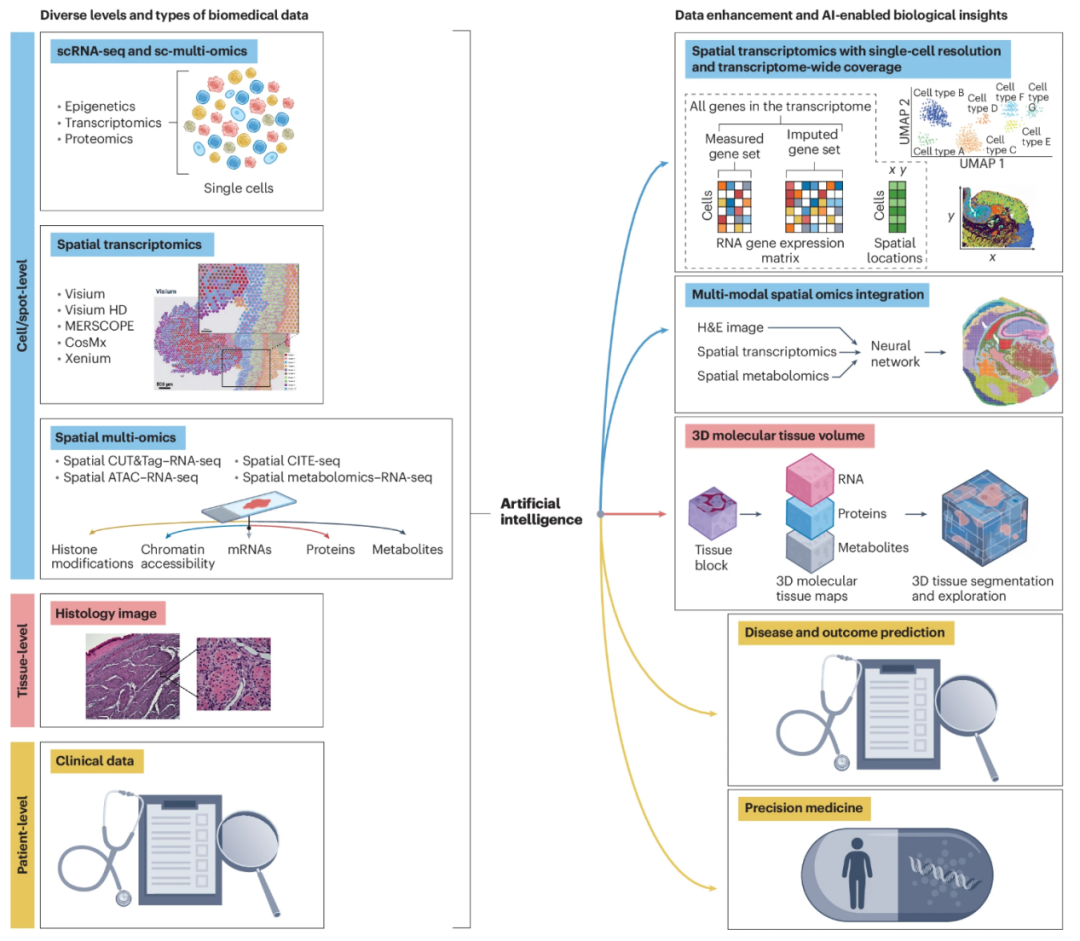
提高空间转录组学的空间分辨率和基因覆盖度
提高空间转录组学的空间分辨率和基因覆盖度
随着Visium HD和Stereo-seq[1]等尖端技术的出现,基于测序的空间转录组学已经达到了在亚细胞分辨率下测量整个转录组中基因表达水平的能力。尽管这种前所未有的精确度能够很好地解析细胞,这些方法仍然很难获得原位单细胞表达谱。由于缺乏细胞边界信息,我们只能获得在一个小区间(Bins)内基因表达谱,这些区间并不对应真实的细胞。因此,特别是在异质性组织切片中,基因表达可能仅代表一个细胞的一部分或包括多种细胞类型的转录本。这一限制阻碍了某些细胞类型的识别,因为其表达量的准确性不足可能会减弱它们的信号强度。此外,对数百万个点进行测序的高昂成本降低了此类超分辨率技术的普及性。相比之下,基于成像的空间转录组学技术,如MERFISH[2]、CosMx[3]和Xenium,能够定位单个转录本并通过细胞分割提供单细胞级别的基因表达谱。然而,这些方法通量不足,每个组织切片最多对数千个基因进行测量,导致空间基因表达的不完整表征。这通常会限制识别罕见细胞类型、表征细胞状态和完全理解复杂的细胞间相互作用的能力。这些局限性给疾病研究带来了挑战。例如,在癌症中,罕见的免疫细胞亚型在对肿瘤的免疫反应中起着关键作用,影响治疗效果和肿瘤行为。因此,准确识别和定位这些罕见细胞类型对于研究肿瘤微环境和发展有效治疗至关重要。因此,实现真实单细胞水平的空间分辨率、全转录组测量仍然是一项迫切需求。
我们认为,空间转录组学中的空白可以通过单细胞RNA测序(scRNA-seq)数据来填补,后者提供了细胞水平上的全转录组测量值,但缺乏空间环境。可通过整合方式有效利用单细胞数据集,具体而言,可以通过使用空间组数据训练深度学习模型,来推断配对的scRNA-seq数据集中细胞的空间坐标,进而使用加工过后的“伪空转数据”完成整合[4]。另一种方法利用scRNA-seq数据集学习基因表达水平之间的关系,并插补基于成像的空间转录组学实验中未测量的基因[5]。我们认为,结合基于成像和基于测序的空间转录组数据和单细胞数据的AI驱动方法可能是生成覆盖整个转录组的全面、单细胞分辨率数据的关键。这些方法还将促进来自细胞图谱项目的不同类型数据的整合,为更广泛的科研社区提供完全集成的数据。
促进空间多组学数据的生成
促进空间多组学数据的生成
最近在空间多组学技术方面的进步使得能够从单一组织切片中获取多种空间解析的组学模式,提供分子组成和组织结构的全面视图[6-8]。然而,提取一种模态的数据可能会损害其他模态所需的分子完整性,从而在后续的分子测序或成像过程中降低敏感度。例如,可以通过先进行MALDI-MSI再进行Visium来同时生成同一组织切片的空间转录组学和代谢组学数据[8]。然而,用于代谢物定位的MALDI-MSI可能导致组织降解,减少可用于测序的RNA分子数量。此外,连续应用针对同一种分子类型的多种测序技术(如CUT&Tag-seq和ATAC-seq,两者都靶向DNA)可能会损害最后测量模态的目标分子的可用性。
为了克服这些局限性,我们认为可以利用AI在计算机上生成空间多组学数据,比实验上的做法可行性更广泛。这涉及从同一块组织中提取多个相邻的组织切片,使用不同的空间组学技术对每一切片进行分子提取识别,同时确保至少有一种锚定模态(如苏木精-伊红染色的组织学图像)贯穿所有切片。然后,对于每一组织切片,可以训练一个AI模型来辨认锚定模态与该切片使用的特定模态之间的关系。利用这些见解,模型可以根据锚定模态建立的模式推断每一组织切片中未观察到的组学模态。此外,利用苏木精-伊红染色组织学图像,可以使用弱监督框架将组织学特征与分子数据结合起来,从而提高空间组学数据的分辨率[9]。随后,组织切片的对齐将形成一个伪三维超分辨率空间多组学数据集,大幅减少了实验生成此类数据集的成本和复杂性。
然而,实施这些AI驱动的方法面临着一些挑战。首先,即使切片来自同一组织块,组织学图像之间往往存在强烈的批次效应,这会妨碍组学预测模型在新组织切片上的有效推广。此外,并非所有分子表达模式都与组织学特征相关联,这限制了这些方法只能推断分子特征的一小部分。而且,空间多组学技术相较于空间转录组学技术还较为不成熟。然而,随着领域的不断发展,我们预计技术突破将缓解这些挑战,最终通过AI驱动的方法使数据更容易为科研界所用。
组织结构的三维重建
组织结构的三维重建
大多数空间组学技术在约5微米厚的单个组织切片的二维空间坐标中捕获分子测量值。虽然这对于理解单个组织切片的分子特性很有价值,但在z轴方向上的缺失限制了从切片的较大组织块中进行生物学发现的能力。这种限制在研究疾病时尤为重要。例如,在癌症中,从肿瘤活检获得的5微米切片仅占整个肿瘤体积的极小部分。这为细胞间的相互作用和肿瘤结构提供了非常有限的视角。此外,因为空间组学技术只能在固定的时间点测量表达,研究者无法直接跟踪肿瘤的发展。因此,他们必须根据现有数据推断肿瘤如何生长和转移。使用单个二维组织切片的空间分子数据进行准确预测几乎是不可能的。因此,我们认为三维空间组学数据对于全面理解肿瘤微环境的复杂性和准确预测肿瘤进展至关重要。
已经有模型用于数字化重建三维组织块,并把连续组织切片对齐到统一的坐标空间中[10]。然而,考虑到有些组织块由数十万份切片组成,全面的空间分子分析所需的巨大成本和时间意味着通常只直接分析其中一小部分。因此,我们认为需要AI方法来构建组织块的完整三维空间分子地图。一种潜在的方法为选定的组织切片生成空间组学和苏木精-伊红(H&E)成像数据,这些切片共同捕捉沿z轴方向存在的结构多样性。如果可以获得剩余组织切片的H&E图像,则可以使用AI方法捕捉分子和组织学特征之间关系,进而来推断未测量切片的空间组学数据[9]。进一步的研究有必要确定用于分析组织切片的最佳数量和选择标准,考虑经济约束和组织块的多样性。
利用多模态大型语言模型增强空间组学
利用多模态大型语言模型增强空间组学
多模态大型语言模型(MM-LLMs),即在大量数据上训练以理解不同模态之间关系的模型,在解决目前空间组学数据生成和分析中面临的许多挑战方面展现出前景。LLMs和MM-LLMs已经在病理成像[11-12]、单细胞RNA测序(scRNA-seq)[13-14]和空间转录组学[15]等相关任务中展示了显著的潜力。通过利用大量的空间组学数据集,我们预计MM-LLMs可用于执行各种任务,如空间域检测、空间可变基因检测和细胞间通讯分析。此外,这些模型能够结合多种组学和医学成像模态,从而提高空间组学数据的分辨率和覆盖度,并推断未测量的组学模态,为本文讨论的许多挑战提供解决方案。此外,我们认为MM-LLMs在精准医学领域具有重要应用前景。具体来说,空间组学技术能够以前所未有的视角揭示疾病相关结构的细胞和分子组织。这些结构及其形成方式对疾病诊断、治疗效果、患者预后以及新型治疗方法的开发具有重要意义。通过关联空间分子模式、病理成像特征与临床结果,治疗方案可以被定制以满足个体患者的独特需求。
展望未来,我们预计可以开发出一种MM-LLM,它将空间组学、医学成像和临床文本数据整合到一个连贯的模型中。该模型最初将在涵盖这些模态的庞大语料库上进行预训练。然后,它将使用更为专业的数据集进行微调,以预测特定结果或根据成像生物标志物和既往病史推荐个性化的治疗计划。诸如Owkin Inc.的MOSAIC项目等举措,旨在跨七种癌症实例生成数千份空间多组学数据集,以识别新的空间生物标志物和患者特异性药物靶标,这样的模型至关重要。随着更多类似MOSAIC的项目完成,越来越多的空间组学数据集将可用于MM-LLM的预训练,而更专业的数据集可用于微调。
结论
结论
尽管近年来在空间组学技术方面取得了显著进展,但我们认为持续存在的局限性凸显了AI在推进这一领域中的核心作用。AI可以促进空间转录组学与scRNA-seq的整合,使我们能够在单细胞水平上测量转录组范围内的空间基因表达谱。此外,通过整合空间组学与组织学成像数据,AI可以构建高分辨率、全面的三维空间组织图谱,覆盖广泛的组学模态。随着可用数据集数量的增长,MM-LLMs可以在空间组学、医学成像和临床文本数据上进行训练,用于生物医学研究和精准医学中的任务。这些模型将加速生物学发现并促进个性化治疗的发展。通过利用AI的力量,我们预计研究人员将解锁空间组学的新维度,带来开创性的发现。
大模型与生物医学:
AI + Science第二季读书会
详情请见:
生命复杂性读书会:
生命复杂系统的构成原理
在生物学中心法则的起点,基因作为生命复杂系统的遗传信息载体,在生命周期内稳定存在;而位于中心法则末端的蛋白质,其组织构成和时空变化的复杂性呈指数式增长。随着分子生物学数十年来的突飞猛进,尤其是生命组学(基因组学、转录组学、蛋白质组学和代谢组学等的集合)等领域的日新月异,当代生命科学临近爆发的边缘。如此海量的数据如何帮助我们揭示宇宙中最复杂的物质系统——“人体”的构成原理和设计原理?阐释人类发育、衰老和重大疾病的发生机制?
集智俱乐部联合西湖大学理学院及交叉科学中心讲席教授汤雷翰,国家蛋白质科学中心(北京)副研究员常乘、李杨,香港浸会大学助理教授唐乾元,北京大学前沿交叉学科研究院研究员林一瀚,中国科学院分子细胞科学卓越创新中心博士后唐诗婕,共同发起「生命复杂性:生命复杂系统的构成原理」读书会,从微观细胞尺度、介观组织器官尺度到宏观人体尺度,梳理生命科学领域中的重要问题及重要数据,由生物学家提问,希望促进统计物理、机器学习方法研究者和生命科学研究者之间的深度交流,建立跨学科合作关系,激发新的研究思路和合作项目。读书会从2024年8月6日开始,每周二晚19:00-21:00进行,持续时间预计10-12周。欢迎对这个生命科学、物理学、计算机科学、复杂系统科学深度交叉的前沿领域感兴趣的朋友加入!
6. 加入集智,一起复杂!
微信扫一扫,分享到朋友圈











