Nature通讯:通过深度生成网络对单细胞转录数据进行双曲嵌入

导语
双曲空间嵌入是研究复杂网络的新兴方法。5月5日一篇发表在 Nature Communications 的一篇论文中,研究者通过结合对抗生成网络对单细胞转录数据进行降维,从而找到特征向量。该文的方法对大规模单细胞和空间组学研究有一定启发。

郭瑞东 | 作者
赵雨亭 | 审校
邓一雪 | 编辑

论文题目:
Deep generative model embedding of single-cell RNA-Seq profiles on hyperspheres and hyperbolic spaces
论文地址:
https://www.nature.com/articles/s41467-021-22851-4
1. 单细胞转录数据降维存在的问题
1. 单细胞转录数据降维存在的问题
通过测量不同类型的单个细胞中,各个基因对应 RNA 的表达量,单细胞转录本测序可以考察不同细胞的异质性,这对生物系统的研究至关重要。由于单细胞测序产生的数据具有虽然维度高,但由于很多处在同一通路上的基因是共表达的特点,其事实的维度很低。同时,由于细胞类型的不同能够解释大部分表达量差异,因此降维已成为单细胞转录本分析中的核心步骤。
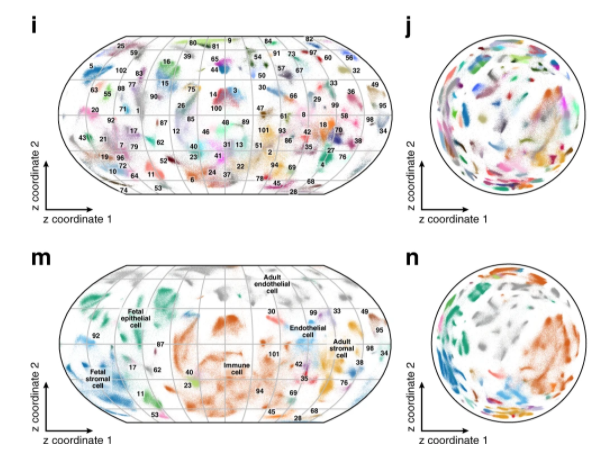
 图1. 使用 tSNE 和 UMAP 两者常用的聚类方法,对102种人类细胞系进行聚类的结果,其中很多不同类型的细胞被聚在一起,且不同类型的细胞间距离更近
图1. 使用 tSNE 和 UMAP 两者常用的聚类方法,对102种人类细胞系进行聚类的结果,其中很多不同类型的细胞被聚在一起,且不同类型的细胞间距离更近
当前基于拓扑的聚类方法——例如常用的 tSNE 和 UMAP、以及单细胞转录组分析的另一类常用方法:变分自编码器(VAEs)——都假设在低维的隐空间上,数据符合多维正态分布。当训练时间足够长后,这会使得来自不同类型的细胞都聚集在隐空间中的高斯分布的中心位置,从而造成不同类型的细胞聚类一起。
单细胞转录组数据分析的第二个挑战,来自当前的模型,都使用余弦距离测量两个细胞之间的表达量差异。当面对不足以分群的单细胞转录组数据(在一个典型的单细胞转录组数据中,超过90% 的基因没有表达)时,经过正则化的余弦距离,等价于欧式距离。然而在稀疏数据集上,欧式距离无法准确描述细胞间的差异,从而对降维工具产生了扭曲。
当前的分析方法,对单细胞转录本的分析需要经过多个独立的步骤:例如正则化、批次校正、数据降维和可视化,每一步都有各自对应的算法,缺少一个端对端的分析框架。同时,每一次的校正,只针对一个单独的因素,例如不同测序平台和批次、不同时间检测等。现有的批次校正方法都无法很好地处理这些由复杂的多重因素造成的影响。但是校正这些混杂因素,对于整合不同研究的数据、解释各种因子对复杂组织中细胞的影响以及最终构建大型的细胞表达谱参考集是至关重要的。
2. 基于深度生成模型的单细胞表达量,
为何需要进行双曲嵌入
2. 基于深度生成模型的单细胞表达量,
为何需要进行双曲嵌入
该文提出的分析框架,名为 scPhere,是一个端对端的模型:不需要对基因表达分布进行正则化和批次校正,就可以捕捉不同细胞间的固有属性、处理复杂的批次效应。该方法通过将表达量矩阵嵌入到低维的双曲球面而非低维欧氏空间,来减少数据处理过程中的扭曲。该方法假设在双曲空间或球形空间中,不同类型的细胞呈均匀分布,因此不会出现不同细胞被迫聚集在一起的情况。
而近来的众多研究,也证明了很多复杂网络对应隐空间中的节点距离,应该是双曲距离。例如:
从复杂网络的视角来看,单细胞转录组的数据降维,就是将表达矩阵这一输入数据中的每一行看成是网络中的节点,找到可以只依赖少数节点,就以相对高的精度重构出整个矩阵的一个生成网络,因此如节点间相互关系,相比欧式距离,双曲距离能以更加不失真、不扭曲的方式描述,这就为使用双曲几何进行建模提供了合理性的理论依据。
传统欧几里德几何学假定平行线之间永远保持相同的距离,既不会相交也不会渐行渐远,而在双曲空间中,平行线会越来越远,一条直线可以有无数条平行线。这导致双曲空间内,面积和体积相对于半径的增长速度要比传统的欧式空间内快的多(指数增长的双曲空间带来了更大的模型容量,使树的节点数目可以随着深度呈指数增长),这使得在双曲空间下,描述网络的几何结构更为便利。
使用双曲几何的另一好处,是方便的三维可视化:只需要旋转球面,将3D空间中任意角度进行投影,就可以进行交互式的可视化,并通过等地球地图投影法(Equal Earth map projection),将三维坐标转换为二维坐标。
3. scPhere 的算法框架
3. scPhere 的算法框架
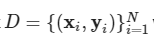
该算法的输入,为一个对应到N个细胞的二元组,其中xi为该细胞的表达谱,yi为批次标签,例如测序发生在细胞发育的某个时间点,其可包含多种不同类型的标签——测序机器、样本来源等。
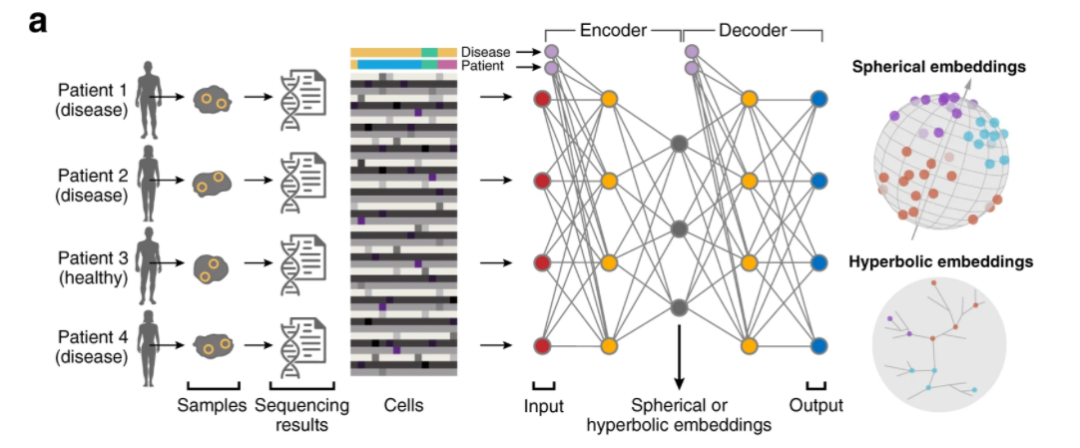 图3. scPhere 的算法流程图,表达量矩阵进自编码器后,对压缩后的特征分别在球形和双曲空间中进行嵌入
图3. scPhere 的算法流程图,表达量矩阵进自编码器后,对压缩后的特征分别在球形和双曲空间中进行嵌入由于 scPhere 是一个基于深度学习的模型,因此其训练过程,可以使用小批量(mini batch)的数据集,执行梯度下降算法来进行训练。该模型特别适合处理包含复杂批次效应的大规模数据集,通过生成与批次无关的低维表征,确定细胞类型,推断细胞在其来源生物样本中所处的位置,并找出细胞间的相互作用,而这都加深了对单细胞转录组数据的理解。
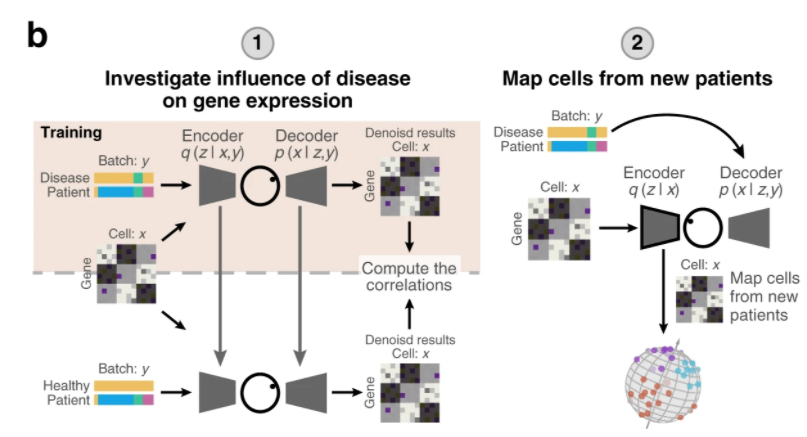 图4. scPhere 可在训练完成后,对新样本生成批次无关的低维表征,找到其对应的球形空间嵌入
图4. scPhere 可在训练完成后,对新样本生成批次无关的低维表征,找到其对应的球形空间嵌入
4. scPhere 在真实数据集上的表现和潜在应用
4. scPhere 在真实数据集上的表现和潜在应用
对于大规模数据集,scPhere 的复杂度,随着输入单元的数量线性增长,该优势意味着即使输入的细胞很多,算法也不会让细胞在聚类时拥挤。在对来自30万个对应不同细胞类型——部分来自患者、部分来自健康人、取自不同地区进行人群的——来自基质、表皮以及免疫细胞的表达谱进行聚类后,可发现左图的 scPhere 中,不同的细胞类型能够清晰地区分开,而常用的 tSNE 和 UMAP ,即使经过了 Harmony批次校正也依然聚在一起。
 图5. 对30万个真实细胞的单细胞转录本数据使用 scPhere、tSNE 和 UMAP 聚类效果对比
图5. 对30万个真实细胞的单细胞转录本数据使用 scPhere、tSNE 和 UMAP 聚类效果对比scPhere 还能够通过找到批次无关的表征,揭示其背后的生物学机制。例如下图所示,左图描述在训练过程中,先只使用来自健康人的、不同类型的成纤维细胞表达量矩阵作为输入,之后使用包含不同类型的炎性成纤维细胞做输入,使用 KNN 基于低维表征预测细胞类型。而右图展示了不同类型细胞的真阳性率,其中训练集中没有出现的炎性成纤维细胞准确性最低;数据集中大部分数据被分类为 WNT2B+成纤维细胞,约10%被分类为 WNT5B+ 成纤维细胞,而这指出了炎症细胞可能的来源。
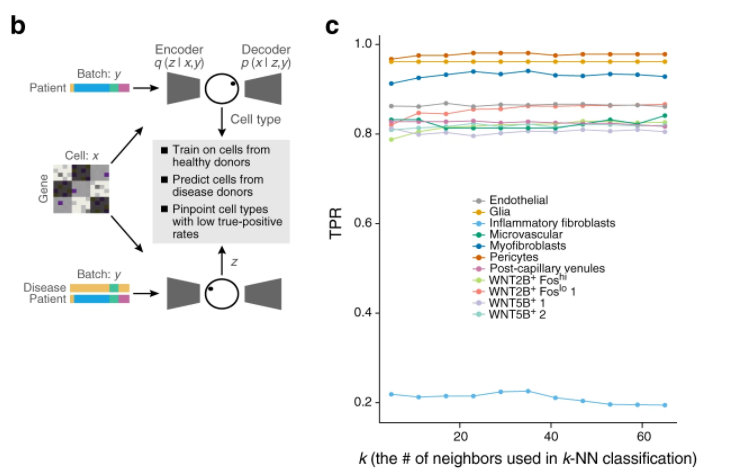 图6. 使用不同类型批次数据训练的 scPhere 的模型进行分类,可揭示隐藏的生物学特征
图6. 使用不同类型批次数据训练的 scPhere 的模型进行分类,可揭示隐藏的生物学特征单细胞分析中,找出对聚类影响最大的标志基因(marker gene)如何在空间分布中变化,可以基于 scPhere 得出的球形空间进行嵌入,推断细胞所在组织的相对空间位置,例如下图展示的,对斑马鱼胚胎外包上半部分的1406个细胞进行单细胞转录组测序,之后对其进行球形空间的嵌入,可以根据特定标志基因的表达量差异(图a),经过 scPhere 推断出细胞所在的空间位置(图b)。为了验证 scPhere 找到的空间特征是稳定的,使用新的来自三个数据集的2802个细胞,绘制了图c和图d,其两者具有相似性。
 图7. 标志基因在球形空间映射后具有批次一致性的嵌入
图7. 标志基因在球形空间映射后具有批次一致性的嵌入
通过双曲嵌入,还可以找到细胞发育随时间变化的情况,下图对比了双曲空间嵌入(Poincare disk)以及欧式空间嵌入下,预估的免疫细胞从干细胞发育的轨迹。可见只有在双曲空间中,够清晰找到不同类型细胞发育中所循序的轨迹。
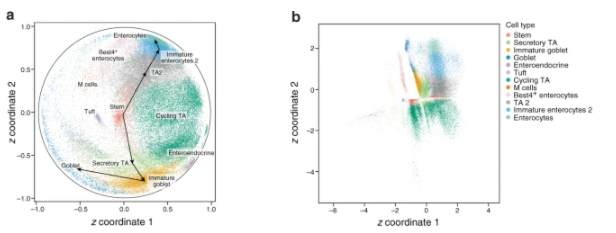 图8. 双曲和欧式空间中免疫细胞的发育轨迹对比
图8. 双曲和欧式空间中免疫细胞的发育轨迹对比
而通过对比线虫不同时间,不同细胞类型的单细胞测序使用 UMAP(下图)和 scPhere(上图)的聚类结果,可以看到不论只是使用采样的胚胎存活时间或者是细胞类型进行标签,scPhere 都能分开并找出不同类型细胞发育轨迹,或将不同时间段的胚胎细胞中对应到连续的表达谱上,这再次论证 scPhere 能够应对多维度的复杂标签,并发现发育过程中的表达量变化轨迹。
 图9. 来自不同时间的线虫胚胎中,不同细胞类型 UMAP 和 scPhere 的聚类可视化对比
图9. 来自不同时间的线虫胚胎中,不同细胞类型 UMAP 和 scPhere 的聚类可视化对比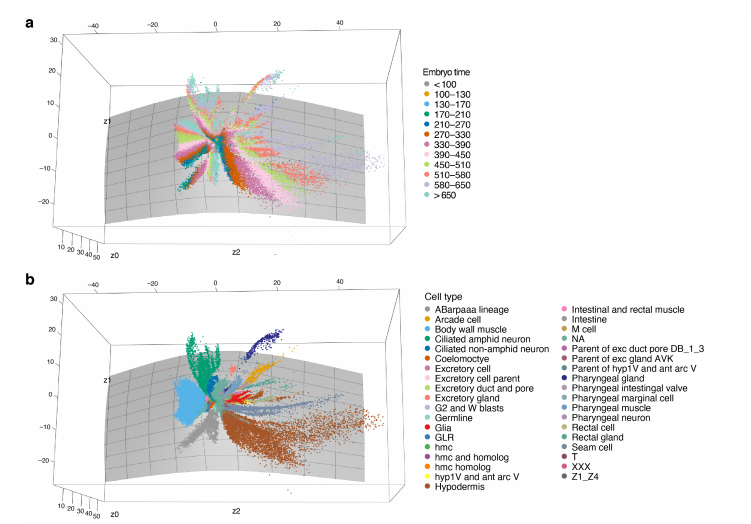 图10. 来自不同时期的线虫胚胎中,不同类型的细胞在三维双曲空间聚类的可视化
图10. 来自不同时期的线虫胚胎中,不同类型的细胞在三维双曲空间聚类的可视化
4. 总结
4. 总结
研究人员对 scPhere 的进一步探索中,一是通过采用半监督学习的方式,基于少量人工标记出类型的细胞,经由双曲嵌入,去为其他类型未知的细胞进行标注。另一方向是扩展 scPhere 对噪音的处理能力,考察其能否应对数据缺失以及处理包含被 RNA 污染的数据集。除此之外,目前缺少高效的工具在双曲空间中进行诸如 KNN 的搜索,这也值得研究。
scPhere 作为一个生成式的单细胞分析框架,其最大优势在于可以对立多维度的复杂批次效应,在校正一个或多个干扰因素影响的同时,对单细胞转录组数据进行可视化和聚类。该方法能量化评估多重干扰因素间任何几个的组合所带来的干扰;调查哪种细胞类型受某个因素(例如状态或位置)的影响最大。
该方法产生的批次无关的低维表征和嵌入,使得经由全新来源、个体或采样条件的数据能够在保留生物学特征的前提下进行映射,从而使得不同来源数的据得以整合,进而构建出细胞表达量参考图谱。鉴于以上三点,以及其在大数据集上的高效处理能力和可扩展性,学界可以预见 scPhere 将是大规模单细胞和空间组学研究的一个有价值的工具。
复杂科学最新论文
集智斑图顶刊论文速递栏目上线以来,持续收录来自Nature、Science等顶刊的最新论文,追踪复杂系统、网络科学、计算社会科学等领域的前沿进展。现在正式推出订阅功能,每周通过微信服务号「集智斑图」推送论文信息。扫描下方二维码即可一键订阅:

推荐阅读
点击“阅读原文”,追踪复杂科学顶刊论文
微信扫一扫,分享到朋友圈











