本文介绍一项由欧洲生物信息学研究所欧洲分子生物学实验室、瑞典斯德哥尔摩大学生命科学实验室以及瑞典斯德哥尔摩大学生物化学与生物物理系联合发表于nature structural & molecular biology的研究工作。作者探究了深度学习方法在预测人类蛋白质相互作用结构上的最新进展以及局限性,探讨了界面残基中潜在的调控机制,并介绍了使用预测的二元复合物构建多聚体的案例,拓展了我们对人类细胞生物学的理解。
蛋白质复合物的结构表征是理解蛋白质功能机制、研究突变影响、研究细胞调控过程的关键步骤。近来,基于神经网络的方法已经具备了能够准确预测单个蛋白质和蛋白质复合物的能力,但其在大规模人类复合物结构预测中的效用尚未得到验证。在此,作者评估了应用AlphaFold2建模大规模人类蛋白质相互作用结构的可能性与局限性。作者预测了65484种来源于不同实验方法的复合物结构,其中3137个预测结构拥有高置信度,它们富集在由实验方法组合支持的预测中,证实了根据置信度评价预测模型的可能性。通过研究界面残基的致病性突变与磷酸化,作者展示了结构上解析相互作用组的价值所在。最后,作者还介绍了使用二元复合物构建多聚体的案例。
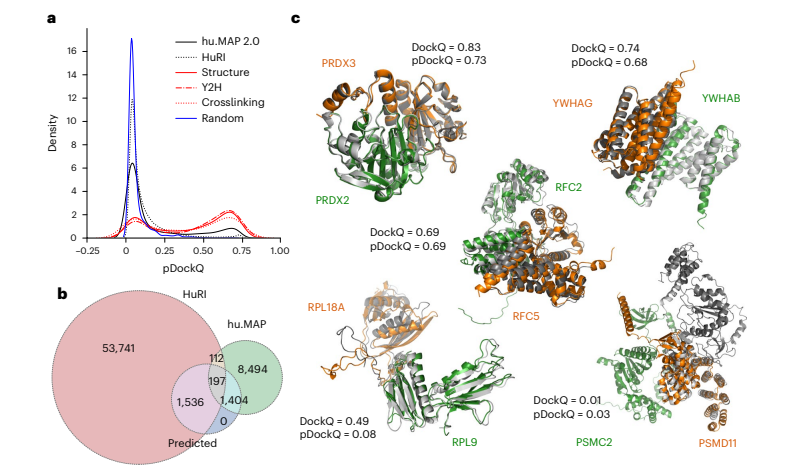
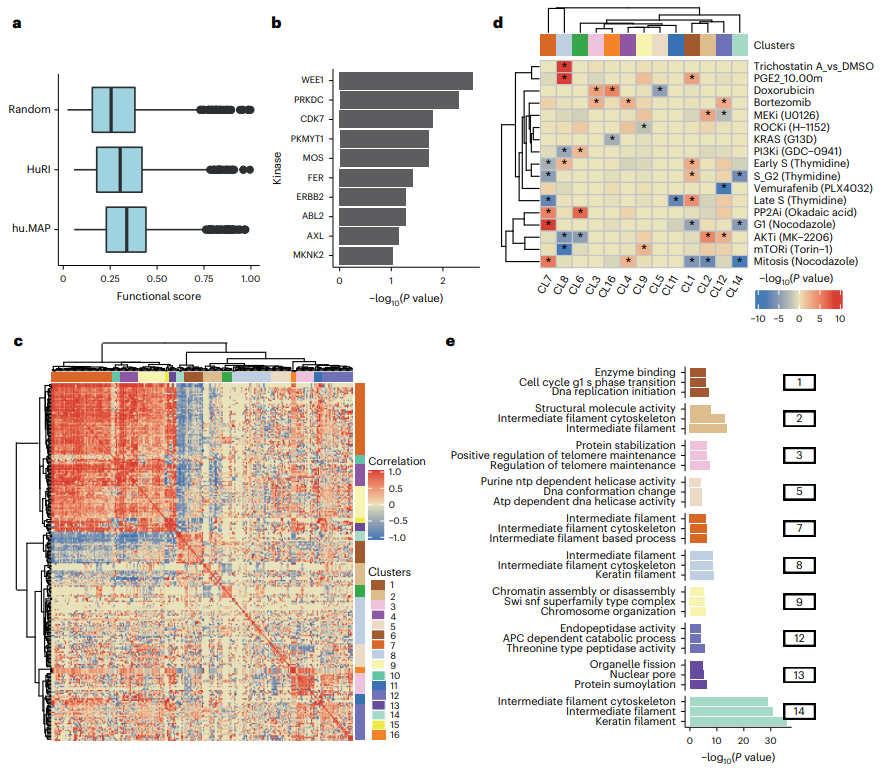
图1. AlphaFold2复合物预测在大规模人类PPIs数据集上的应用
作者使用了基于AlphaFold2的FoldDock管线,对65484条来源于HuRI与hu. MAP v.2.0数据库的实验测定的人类蛋白质相互作用结构进行预测。如同FoldDock管线中一样,作者将界面的尺寸与plDDT分数组合成pDockQ用来预测复合物的DockQ分数,它可以根据置信度对模型进行排序。在图1中,作者展示了不同场景下蛋白质相互作用的pDockQ分数分布。结果显示,已知相互作用的蛋白质的pDockQ分数往往高于随机集合,并且对于hu. MAP数据集预测的置信度高于HuRI数据集,这表明高置信度的模型富集在具有高亲和力和直接相互作用的蛋白质相互作用区域。此外,作者还展示了与实验模型或同源模型对齐的预测结构示例,阐述了预测与置信度评分如何与观测到的对齐相联系起来。总之,实验结果表明AlphaFold2可以预测大型复合物中直接相互作用的蛋白质对的结构。
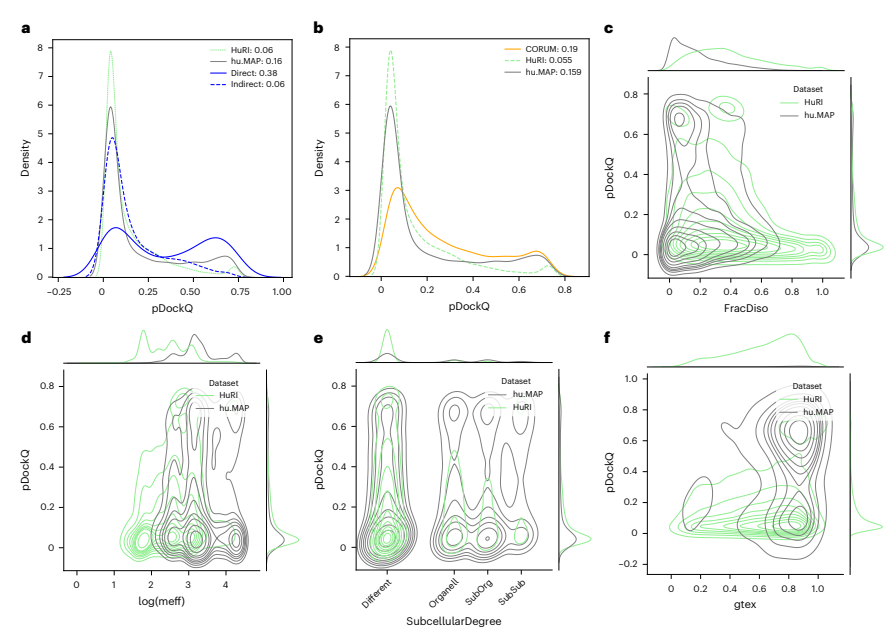
图2. 影响预测置信度的蛋白质和相互作用特征:不同数据集的分析
如图1所示,相较于HuRI与hu. MAP数据库中的蛋白质对,出现在PDB库中的蛋白质对更加富集于高分模型部分。为了更好的理解这种差异,作者首先研究了一个由大型异质蛋白复合物构成的额外数据集,其中的蛋白质对可以根据是否具有直接相互作用进行划分。预测结果表明,直接相互作用对与间接相互作用对之间pDockQ分数的差异是显著的,这表明即使直接相互作用对是大型复合体的一部分,也往往能够被预测。为了探究hu. MAP与HuRI数据库预测置信度之间产生差异的原因,作者引入了CORUM数据库作为对照,实验结果表明不同的蛋白质复合物数据库之间高置信度预测的比例相似,而HuRI数据库是个例外,这可能是因为HuRI数据库中的许多蛋白质相互作用是瞬时的,AlphaFold2恰巧不能有效地预测这种相互作用。
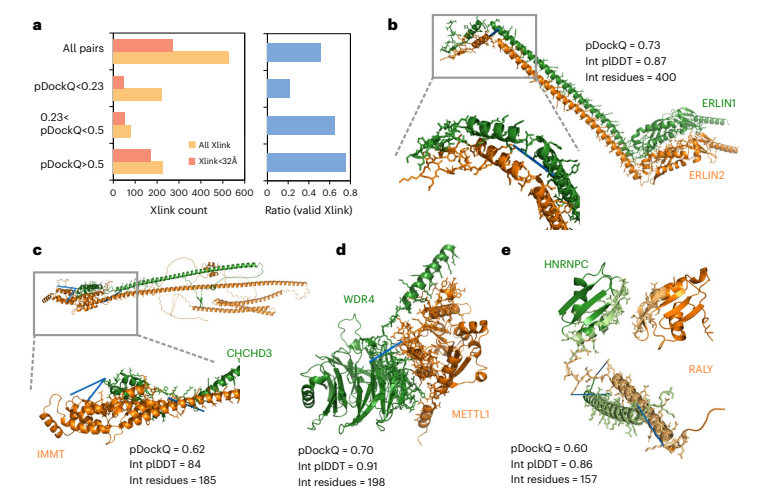
化学交联质谱是一种可用于识别附近反应性残基的方法,在一对蛋白质中识别此类残基可以辅助定义可能的蛋白质界面。为了确定预测的复合物结构是否满足这种正交空间约束,作者获取了528个具有预测模型的蛋白质对的残基对交联集合。其中,51%的模型在低于预期最大距离的距离处有一个或多个交联。当把pDockQ分数上调,将预测模型的置信度限制在更高水平时,具有可接受交联的复合物的比例也随之增加,在pDockQ分数大于0.5时,其比例达到了75%,这与基准测试结果一致。作者随后在图3中提供了多个案例说明了化学交联验证的有效性。
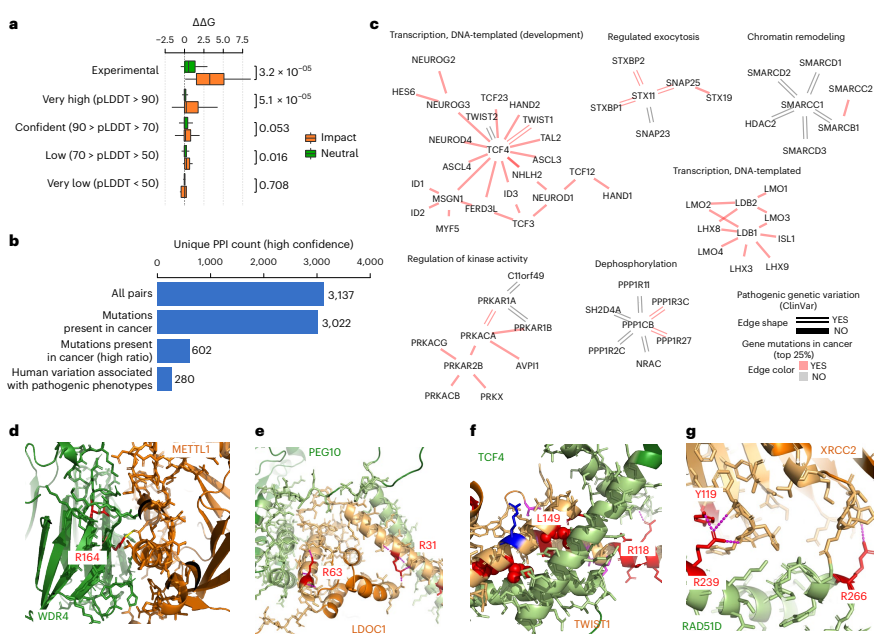
为了确定预测结构的有用性,作者整理了一组位于界面残基上的突变,这些突变已经通过实验测试了对相应相互作用的影响。作者在这些突变上使用FoldX对突变的结合亲和力变化进行了预测并观察到破坏相互作用的突变强烈影响了结合的稳定性。更进一步的实验证实,在使用FoldX力场估计突变结合亲和力的影响时,只有非常精确的模型是有效的。随后,作者将人类疾病和癌症突变映射至由高置信度蛋白复合物预测集所定义的界面残基中,发现相对于蛋白质的其他部分,在界面残基上的致病突变与良性突变具有很强的富集。最后,在图4中,作者就具有界面残基突变的蛋白质网络簇和具有疾病突变的界面残基进行了举例说明。
将磷酸化位点的位置映射到蛋白质界面可以为其在控制蛋白质相互作用中所扮演的角色提供机制假设。作者利用最近对人类磷蛋白的描述,在高度可信的模型中,在界面残基处鉴定出4145个独特的磷酸基,并发现位于界面处的磷酸化位点较随机位点来说具有更高的功能重要性,且某些界面可能收到特定激酶和条件的协调调节。之后,作者深入研究并举例论证了潜在的共同调节界面。虽然并不是所有的磷酸盐都可能调节结合亲和力,但该分析为多种蛋白质在特定扰动后通过调节相互作用来协调调节提供了假设。
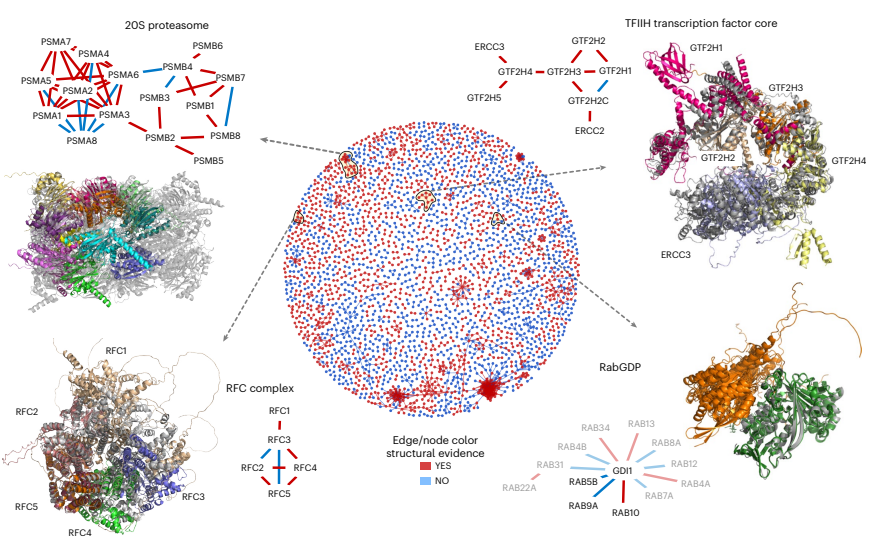
蛋白质既可以同时与多个蛋白相互作用,作为更大的蛋白质复合物的一部分,又可以在时间和空间上分离。这也反映在了作者提出的结构特征网络中,即蛋白质可以被分组发现,正如蛋白质相互作用全局网络视图中所示。由于计算资源的限制,作者仅在该网络覆盖的一小组复合物上测试了迭代结构对齐的过程,并将实验确定的结构与预测模型对齐,分析了该过程的潜力与局限性。伴随着测试示例的鼓舞,作者设计了一个自动化的流程,通过迭代对齐的方式生成更大的复合物,在证实其可行性的基础上进一步分析了限制这一过程成功的一些因素。
作者通过一系列实验分析了使用AlphaFold2进行蛋白质复合物预测的可行性,并注意到数据来源对预测的效果具有重要影响。分析表明,结合基于亲和力、共分离技术、互补的方法支撑的蛋白质相互作用可以产生更高置信度的模型。作者提出,可以使用来自模型的评估指标对高置信度模型进行排名,为大规模PPI和稳定复合物的研究提供帮助,而交联质谱实验数据为进一步验证这些想法提供了理想的资源。此外,对界面残基错义突变以及磷酸化的深入分析也有利于对分子机制的进一步理解。文末,作者更是提出了从二元复合物出发建立更大复合物结构模型的想法,并对其潜力与局限予以分析,为构建多聚体提供了广阔的思路。
Burke, D.F., Bryant, P., Barrio-Hernandez, I. et al. Towards a structurally resolved human protein interaction network. Nat Struct Mol Biol (2023).
https://doi.org/10.1038/s41594-022-00910-8
集智斑图顶刊论文速递栏目上线以来,持续收录来自Nature、Science等顶刊的最新论文,追踪复杂系统、网络科学、计算社会科学等领域的前沿进展。现在正式推出订阅功能,每周通过微信服务号「我的集智」推送论文信息。扫描下方二维码即可一键订阅: