关键词:深度学习,神经网络,分子动力学模拟,原子模型
论文题目:Learning local equivariant representations for large-scale atomistic dynamics
论文来源:Nature Communications
论文链接:https://www.nature.com/articles/s41467-023-36329-y
分子动力学(MD)和蒙特卡洛(MC)模拟方法是计算化学、材料科学和生物学的一个核心支柱。从能源材料到蛋白质折叠的各种应用的共同要求是,势能和原子力的预测必须既准确又有计算效率,以便忠实地描述复杂系统在长时间尺度上的演变。对分子和材料的势能面进行同时精确和高效的计算参数化,是自然科学的一个长期目标。虽然以原子为中心的信息传递神经网络(message passing neural networks, MPNN)已经显示出非凡的准确性,但其信息传播限制了可获得的长度尺度。相反,局部方法可以扩展到大型模拟,但精度较低。
这项工作介绍了 Allegro,这是一个严格的局部等变的深度神经网络原子间势架构,同时表现出卓越的准确性和可扩展性。Allegro 使用学习到的等变表征的迭代张量积来表示多体势,而无需以原子为中心的信息传递。Allegro 在 QM9 和 revMD17上获得了对目前最先进方法的优势。在QM9上,单个张量乘积层的性能超过了现有的深度信息传递神经网络和转换器。此外,Allegro 对分布外的数据显示出显著的泛化能力。使用 Allegro 进行的分子模拟恢复了非晶电解质的结构和动力学特性,与人工模拟非常一致。最后,作者还用1亿个原子的模拟来演示并行化。
总之,Allegro 方法在综合准确性和可扩展性方面,超越了以原子为中心的原子间相互作用的消息传递神经网络模型。这使得从数百万原子的复杂系统的分子动力学模拟中预测结构和动力学特性成为可能,几乎是第一原理的保真。
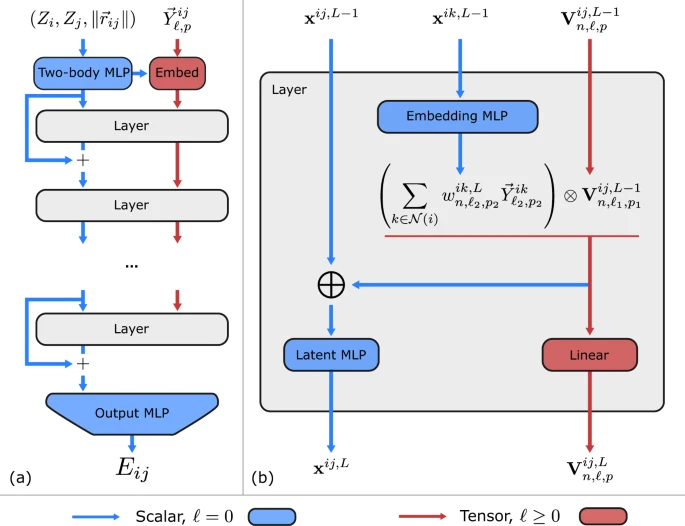
图1. Allegro 网络。(a)展示了 Allegro 模型架构,(b)详细描述了张量积层。蓝色箭头和红色箭头分别代表标量和张量信息,⊗表示张量积,⊕ 是级联。
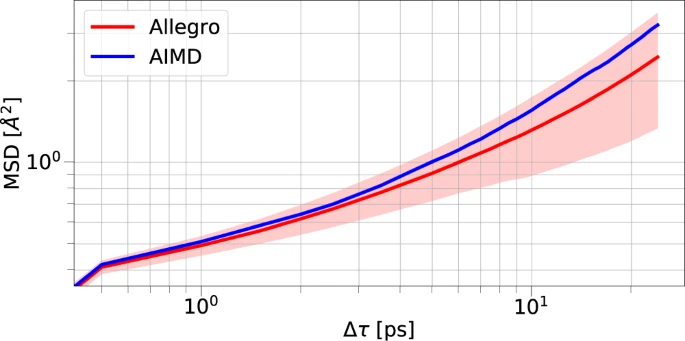
图2. Li3PO4中的锂动力学,AIMD 与 Allegro 的 Li MSD 比较。
集智斑图顶刊论文速递栏目上线以来,持续收录来自Nature、Science等顶刊的最新论文,追踪复杂系统、网络科学、计算社会科学等领域的前沿进展。现在正式推出订阅功能,每周通过微信服务号「我的集智」推送论文信息。扫描下方二维码即可一键订阅: